


Summary
Vi beskriver de billeddannende metoder, vi bruger til at undersøge fordelingen og mobilitet for de transficerede fluorescerende proteiner er hjemmehørende i det endoplasmatiske reticulum (ER) ved hjælp af konfokal billedbehandling af levende celler. Vi har også ultrastrukturelt analysere effekten af deres udtryk på arkitektur i dette subcellulære rum.
Abstract
Lipider og proteiner i eukaryote celler kontinuerligt udveksles mellem cellekamre, selv om disse bevarer deres karakteristiske sammensætning og funktion til trods for intense interorganelle molekylære trafik. De er beskrevet i dette dokument teknikker er effektivt middel til at studere protein og lipid mobilitet og handel in vivo og i deres fysiologisk miljø. Fluorescens opsving efter fotoblegning (FRAP) og fluorescens tab fotoblegning (FLIP) er meget udbredt live-cell imaging teknikker til at studere intracellulær menneskehandel gennem exo-endocytiske proces, kontinuiteten mellem organeller eller underkamre, dannelsen af protein-komplekser, og protein lokalisering i lipid mikrodomæner, kan alle blive observeret under fysiologiske og patologiske tilstande. Begrænsningerne ved disse fremgangsmåder er hovedsageligt på grund af anvendelsen af fluorescerende fusionsproteiner, og deres potentielle ulemper omfatter kunstig overudtrykkerion i celler og mulighed for forskelle i foldning og lokalisering af mærkede og native proteiner. Endelig er som grænse for løsningen af optisk mikroskopi (omkring 200 nm) ikke tillader undersøgelse af den fine struktur af ER eller de specifikke underkamre der kan stamme i celler under stress (dvs. hypoxi, medicin administration, overekspression af transmembrane ER hjemmehørende proteiner) eller under patologiske tilstande, vi kombinerer live-cell imaging af dyrkede transficerede celler med ultrastrukturel analyser baseret på transmission elektronmikroskopi.
Introduction
Opdagelsen af grønt fluorescerende protein (GFP) og dets spektrale varianter, og den parallelle udvikling af fluorescens mikroskopi, har åbnet helt nye muligheder for undersøgelse af protein adfærd i celler. Teknikker, såsom fluorescens opsving efter fotoblegning (FRAP) og fluorescens tab fotoblegning (FLIP), der er mulig på grund af den iboende evne fluorophores at slukke deres fluorescens under intens belysning, er baseret på konfokal live-cell imaging og brugen af transficerede fluorescerende fusionsproteiner 1-3. De er meget udbredt til at vurdere ikke kun lokalisering af proteiner, men også deres mobilitet og vesikulær transport, som kan afsløre vigtige spor vedrørende deres funktion 4.
Det unikke i eukaryote celler, er tilstedeværelsen af intracellulære rum, der har specifikke lipid-og proteinpræparater. Selvom organeller er fysisk isolated, de har brug for at kommunikere med hinanden og udveksle molekylære komponenter for at opretholde cellulær homeostase. Udskillelsen garanterer, at proteiner og lipider syntetiseret i ER nå den korrekte endelige bestemmelsessted, hvor de udøver deres funktion. Intracellulære organeller kan også tilsluttes med dynamiske kontakt sites, der tillader molekyler (lipider), der skal udveksles direkte mellem rummene. Desuden har mange proteiner samles i store heteromere komplekser eller forbundet med specifikke lipid arter (lipidklumperne / mikrodomæner) for at blive funktionelt aktive eller at blive transporteret til deres endelige bestemmelsessted. Alle disse biologiske aspekter stor indflydelse de kinetiske egenskaber af proteiner, og de kan derfor hensigtsmæssigt undersøges ved hjælp af de nedenfor beskrevne teknikker.
Vores gruppe har udbredt FRAP og Flip kombineret med elektronmikroskopi for at studere arkitektur ER og dens forskellige subdomæner. ER er den første station i sekretionsvejen og spiller en central rolle i protein og lipid sortering 5.. Det er en meget dynamisk organel hvis distinkt subdomæner afspejler dens mange forskellige funktioner (dvs. protein og lipid biosyntese og menneskehandel, protein foldning, Ca 2 + opbevaring og frigivelse, og miljøfremmede metabolisme). Men selv om de er morfologisk, rumligt og funktionelt forskellige, disse domæner er kontinuerlige med hinanden og deres relative overflod kan ændres i celler under fysiologiske og patologiske tilstande. Den bedst kendte og normalt rumligt adskilte domæner af ER er den nukleare kuvert, og den glatte og ru ER, men vi og andre har vist, at der er ER-strukturer med en mere omfattende arkitektur og tre-dimensionelle organisation i forskellige celletyper og væv under fysiologiske betingelser, som også kan induceres ved hjælp af stressende stimuli, såsom hypoksi, narkotikaadministration eller overekspression af ER-resident transmembrane proteiner 2,6 (og referencer deri).
Vi har også for nylig vist tilstedeværelse af sådanne strukturer i celle modeller for humane sygdomme 1,7. Stammer fra det stablede cisternae af glatte ER, fik de den kollektive navn organiserede glatte endoplasmatiske reticulum (OSER) i 2003 6, selv om de også er kendt som karmellae, lameller og krystalloide ER på grundlag af deres arkitektur, der ligesom deres størrelse, kan variere. Efter at cellerne transficeres med GFP fusioneret til den cytosoliske region hale forankret (TA) ER-resident proteiner (d EGFP-ER), den svagt dimeriserende tendens GFP i trans dramatisk ændrer organisation og struktur ER. FRAP og FLIP forsøg viste, at d EGFP-ER er fri til at diffundere i OSERs, og at den bevæger sig fra det retikulære ER til OSER og omvendt </ Em> viser, at aggregaterne er sammenhængende med den omgivende retikulære ER. Ultrastrukturel analyse har tilladt os at korrelere fluorescens data med en detaljeret beskrivelse af OSER arkitektur og organisation på nanoskala niveau: OSERs altid består af stakke af parrede cisternae af glatte ER, men kan have forskellige former for fysisk planlægning, såsom regelmæssigt arrangeret sinusformet arrays eller hvirvler eller sekskantede "krystalloide" rørformede arrays. Disse omrokeringer føre til kubiske morfologier 8, der, som de er blevet fundet i cellerne under fysiologiske betingelser 9 og følgende belastninger såsom hypoksi 10, narkotikabehandling 11, og kræft 9, kan have et betydeligt potentiale som ultrastrukturelle markører.
Efter denne første demonstration ved hjælp af GFP fusionsproteiner brugte vi billeddannelse eksperimenter til at analysere udbredelsen af ER-domæner som reaktion på farmakologiske behandlinger 12, Assess tendens fluorescerende proteiner til oligomerise i celler 13, og for at undersøge, hvilken rolle af en mutant ALS-linked TA protein i dannelsen af intracellulære aggregater af ER oprindelse, der kan være relevante for dens patogenicitet 1,8. Det er blevet foreslået, at dannelsen af intracellulære aggregater (som forekommer i mange neurodegenerative sygdomme, 14) kan være en beskyttende mekanisme for at forhindre interaktioner mellem toksiske mutantproteiner og de omkringliggende cellebestanddele 15.
Hvad der følger er en beskrivelse af en kombination af optiske og elektronmikroskopi metoder til at undersøge konstruktioner hvis C-terminale hydrofobe domæner er indsat i membranen af ER, og en analyse af deres dynamiske adfærd og virkningerne af deres overekspression på ER domæne arkitektur i dyrkede celler (se figur 1 viser et flowdiagram af den eksperimentelle protokol).
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1.. Plasmid Celledyrkning og transfektion med ER fluorescerende proteiner
- Plasmidet anvendt i denne undersøgelse består af en forbedret version af GFP fusioneret ved sin C-terminus til haleregionen af ER-isoform af rotte-cytochrom b (5) (forkortet her som b) (5) via en linkersekvens. Haleregionen indeholder hele sekvensen (Pro94-Asp134), som forbliver membranassocieret efter trypsin-spaltning af nativ b (5), herunder 17-rest TMD (transmembrant domæne), flankeret af opstrøms og nedstrøms polære sekvenser (UPS og DPS) . Linkeren består af myc-epitopen, efterfulgt af [(Gly) 4 Ser] 3, og hele cDNA indsat i Hind3-Xba1 sider af den mammale ekspressionsvektor pCDNA3. Detaljerne i konstruktionen af dette plasmid er blevet beskrevet i en tidligere publikation, hvori det er nævnt som GFP-ER 16.
- Grow COS-7-celler i Dulbeccos Modified Eagle Medium (DMEM) suppleret med 10% fetal bovint serum, 2 mM L-glutamin, 1% penicillin / streptomycin i en inkubator ved 37 ° C og 10% CO2.
- Transfektion. Plate 3 x 10 5 celler på en runde dækglas i en 6-brønds plade, og på den følgende dag, transficere med JetPEI som beskrevet af fabrikanten. Bemærk, at den optimale JetPEI / DNA-forhold er blevet testet for at fastslå den maksimale transfektionseffektivitet afhængigt af plasmidet og cellelinje: i vores tilfælde, en JetPEI: DNA-forhold på 2:1 fører til 70-80% transfektionseffektivitet.
2. Levende Fluorescens konfokal mikroskopi
- Live-cell imaging. Sæt dækglasset, som de transficerede celler blev podet i en stål kultur cellekammer 24 mm dækglas fyldt med DMEM w / o phenolrødt suppleret med 10% FBS, 2 mM L-glutamin, 1% pen / strep, 25 mM HEPES 50 pg / ml cycloheximid og 1:100 OxyFluor at forhindre prøver fra fotoblegning. En SP5 konfokal microscope er udstyret med en temperatur-kontrolleret CO 2 inkubator (37 ° C og 5% CO 2) anvendes til levende celler billeddannelse eksperimenter med d GFP-ER bliver visualiseret ved hjælp af en 488 nm laser og en 525/50 band pass emission filter.
- Fluorescens opsving efter fotoblegning (FRAP). Tegn en region af interesse (ROI), svarende til en OSER struktur, og blege det ved hjælp af 20 gentagelser og en kombination af 488 nm (100% af en 30 mW Argon laser, svarende til 5,5-6 uW på prøve) og 405 nm (60% af en 30 mW Diode 405 laser, svarende til 11,6 uW ved prøven) lasere, som efter vores erfaring, fører til effektiv og hurtig fotoblegning.
- Optag inddrivelse af fluorescens i de bleget ROIs ved at tage et enkelt billede hver 10 sek i 10 min (pixel tid = 1,61 usek / px).
- Fluorescens tab fotoblegning (FLIP). Tegn en ROI svarende til en OSER struktur og blegemiddel som beskrevet ovenfor. Blegningen gentages hvert 30. sek,og post-blegning billeder optages hvert 10. sekund i 30 min (pixel tid = 1,61 usek / px).
- FRAP og FLIP analyse. Alle billeder analyseres ved anvendelse ImageJ software ( http://rsbweb.nih.gov/ij/download.html ). I FRAP forsøg fluorescensen genopretning af den blegede ROI målt over tid og normaliseret til den samlede fluorescens bleget cellen, hvilket kontrolleres altid at være konstant over tid.
- For Flip eksperimenter, tegne en ROI uden for bleget OSER og dækker hele cellen. Måle dens fluorescensintensitet med tiden og normalisere de niveauer af fluorescens i et ROI trukket på et ubleget celle med henblik på at korrigere for eventuelt fald i fluorescens forårsaget af billeddannelse selv.
- I alle forsøgene subtraheres baggrunden signal (bestemt i et område uden for celler), fra de fluorescerende intensiteter af ROI'er. Endelig plotte resultaterne ved hjælp af GraphPad Prism software.
3. Ultrastrukturelle Analyse ved Transmission Electron Microscopy
Da toksiciteten af mange af de reagenser og alle de procedurer, der skal udføres iført en passende laboratorium. frakke og handsker under en emhætte.
- Efter fjernelse af dækglas fra petriskålen løse de resterende celler dyrket på bunden af skålen som et monolag ved anvendelse af filtreret 2% glutaraldehyd i 0,1 M cacodylatbuffer, pH 7,4, i 10 minutter ved stuetemperatur.
- Skrabe celler under anvendelse af en Teflon-skraber og overføre dem i 1,5 ml Eppendorf-rør. Pelletere cellerne ved hjælp af centrifugering ved 9.000 g i 10 min. Supernatanten fjernes, tilsættes frisk fikseringsvæske og lade natten over ved 4 ° C.
- Vask pellets buffer og derpå efter-fix med en opløsning af 1% osmiumtetroxid i cacodylatbuffer i 1 time ved stuetemperatur.
- Skyl med MilliQ vand, og en bloc pletten med1% uranylacetat i destilleret vand i mellem 20-60 min.
- Dehydrer prøverne i stigende ethanol-serien (70%, 80%, 90%, 100% og 100% i 10 min hver), og vask kortvarigt to gange i propylenoxid (15 min hver).
- Infiltrere prøver i en blanding af propylenoxid + Epon (1:1) (fra 2 timer til natten over).
- Integrere i Epon epoxyharpiks hærdet ved 60 ° C i mindst 24 timer.
- Afsnit de manuelt trimmede harpiks blokke ved hjælp af en ultramikrotom LEICA UC6 udstyret med en 45 ° diamant kniv til at opnå sektioner med en tykkelse på 60-70 nm. Saml afsnittene om 300 mesh kobber net.
- Farv afsnittene på gitteret med en mættet opløsning af uranylacetat (20 min) og blycitrat (7 min), vaskes grundigt tavlerne ved at nedsænke dem i bi-destilleret filtreret vand, og lad dem tørre ved stuetemperatur.
- De farvede gitre observeres ved hjælp af en Tecnai G2 transmissions elektron mikroskop, og billederne er taget med et bottOM-monterede CCD-kamera på forskellige endelige forstørrelser (mellem 6,000-39,000 X).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Figur 2 viser et eksempel FRAP undersøgelse af protein mobilitet. Mobiliteten for d EGFP-ER protein demonstreret ved den hurtige fluorescens opsving efter fotoblegning i bleget OSERs. For den kvantitative analyse blev den halve tid og mobil fraktion stammer fra eksperimentelt målte data ved at montere følgende monoeksponentiel ligning:
F (t) = F indlæg + (F rec-F post) (1-e-t / τ)
hvor F indlæg er fluorescens signal efter fotoblegning, F rec er den maksimale fluorescens opsving værdi, der nå efter blegning, t tidspunktet for registrering og τ den tid konstant.
Bemærk vigtigheden af at erhverve billeder uden mættede pixels, der kunne ændre fluorescens opsving og dermed den analyse protein mobilitet. Det jeger også vigtigt altid normalisere fluorescenssignalet i den blegede ROI til den samlede fluorescens af den samme celle med henblik på at overveje fluorescensintensitet variationer på grund blegning under billedet erhvervelse eller små ændringer i fokusplan.
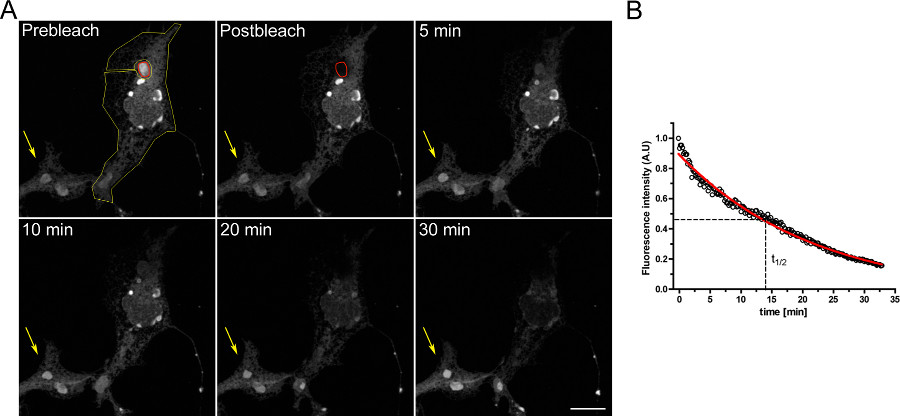
Et eksempel på en FLIP eksperiment for at undersøge kontinuitet mellem intracellulære rum er vist i figur 3.. OSERs er fysisk forbundet med resten af ER som det fremgår af den gradvise tømning af ER, når OSER domænet kontinuerligt bleget.
For en grundig analyse må køb af mættede pixels undgås (se ovenfor), desuden skal erhvervelse parametre sættes op med laser beføjelser så lave som muligt for at undgå fotoblegning grund billede erhvervelse. Af denne grund er det stærkt anbefales at billedet en ubleget celle i samme felt, der skal bruges til at normalisere fluorescenssignal af bleget cell.
Alle forsøg skal udføres i nærvær af cycloheximid, en oversættelse inhibitor, for at undgå en stigning i ER fluorescenssignal (og dermed totale fluorescens) på grund af protein-biosyntese.
Transmissionselektronmikroskopi viste, at de fluorescerende aggregater observeret i dyrkede celler transficeret med d EGFP-ER repræsenterer pletter af glatte og fladtrykt ER cisternae der rumligt organiseret sig i veldefinerede 3D geometrier er klassificeret på grundlag af deres mønstre: lineære eller buede stakke (ofte associeret med kernemembranen ikke vist) (figur 4A og B), der kan være kontinuerlig med regioner med sinusformet ER (figur 4A), membranerne i nogle regioner er organiseret i gitre med kvadratisk eller sekskantet symmetri (krystalloide ER ikke vist ). Tilstødende cistemae er adskilt af et tyndt lag af svagt elektrontiltrækkendetætte cytoplasma omkring 11 nm tykt, der er kontinuert med cytoplasmaet omkring aggregater.

Figur 1. Flow diagram af den eksperimentelle procedure. De dyrkede celler først transficeres med jetPEI protokollen (se) for at over-udtrykke fluorescerende fusionsprotein af interesse. Efter 24 timer er de levende transficerede celler visualiseret FRAP og Flip eksperimenter udføres ved hjælp af en konfokal mikroskop udstyret med en kontrolleret temperatur CO 2 inkubator, og de optagede billeder eksporteres og analyseres ved hjælp af passende software (f.eks ImageJ). For ultrastrukturelle analyse, bliver de transficerede celler faste, pelleteret og indlejret i Epon epoxy harpiks blokke. Ultratynde sektioner er opnået ved anvendelse af en diamantkniv, indsamlet på COpper gitre og observeret under et transmissions elektron mikroskop. Klik her for at se større figur .

Figur 2. FRAP forsøg under anvendelse af COS-7-celler transient transficeret med d EGFP-ER. A) To Oser strukturer (rød ROIs) blev bleget, og fluorescens opsving blev registreret over tid. Klar fluorescens opsving kan påvises 1 min post-blegning, og signalet yderligere stigninger 4 min senere (skala bar 10 um) B):. Kvantitativ analyse af FRAP eksperiment viser inddrivelse halv tid og den mobile del af d EGFP -ER-protein. Click her for at se større figur

Figur 3. FLIP eksperiment ved hjælp af COS-7-celler transient transficeret med d EGFP-ER. A) Den kontinuerlige blegning af en OSER (angivet med røde ROI) forårsager en progressiv nedgang i fluorescens i resten af ER og andre Osers strukturer i den samme celle (angivet af den gule ROI). Den gule pil viser en del af en ubleget celle, hvori fluorescenssignalet er konstant over tid. (Skala bar 10 um). B) kvantitativ analyse af FLIP eksperiment. Klik her for at se større figur
Figur 4.. Efter fiksering og forankring blev celler, der udtrykker, hvor Oser strukturer kunne påvises ved hjælp af fluorescens-optiske mikroskopi høje niveauer af d EGFP-ER observeret gennem et transmissions elektron mikroskop. A) lav forstørrelse af en del af en celles cytoplasma indeholder en OSER bestående af stablede cistemae og bølgende sinusformede membraner. Mitokondrier (M) kan ses samlet omkring de Oser strukturer, mens ribosomer dekorere kun membraner i yderste cisternae (pilespidser og indsat). Den 11 nm tyk elektron-tætte rum mellem membranerne er kontinuert med cytoplasmaet (pil og indsat) (L = lysosomer / (auto-) phagosomes) (skala bar 1,5 m; indsat 0,25 um). B) En OSER kan dannes ved lamellær ER: dvs stakke af fladtrykt ER cisternae der kan være kontinuerlig eller fragmented i deres udseende i tynde sektioner. Vesikler spirende fra den yderste cisternae af stakken kan lejlighedsvis iagttages (stjerne) (PM, plasma membran) (Skala bar 150 nm).
Subscription Required. Please recommend JoVE to your librarian.
Discussion
De protokoller og billedteknik, der er beskrevet i dette papir er blevet brugt til at undersøge fordelingen og mobilitet transficerede TA fluorescerende proteiner er hjemmehørende i Skadestuen af levende celler. Vi har også analyseret virkningen af over-ekspression af disse proteiner på arkitektur i dette subcellulært afsnit ved hjælp af ultrastrukturelle analyser.
Kombinationen af live-cell konfokal billedbehandling og elektronmikroskopi repræsenterer er en meget kraftfuld måde at efterforske de dynamiske egenskaber af proteiner, og kan give vigtige oplysninger om protein funktion. De beskrevne metoder ikke er tidskrævende (typisk tre dages arbejde), og udviklingen af mange brugervenlige programmer for indsamling og analyse billede gør fotoblegning-baseret, live-cell imaging forholdsvis enkel.
Den største begrænsning af disse teknikker er anvendelsen af fluorescerende fusionsproteiner fordifluorescerende mærke kan påvirke den korrekte foldning og / eller samling af proteinet af interesse. Derudover kan overekspression ændre adfærden af transficerede, fluorescens mærkede proteiner, og kan derfor ikke afspejle de reelle egenskaber endogene proteiner, men dette kan overvindes ved anvendelse af inducerbare og stabilt transficerede celler, i hvilke ekspressionsniveauet kan være netop moduleret at opnå niveauer kan sammenlignes med de af det endogene protein 1,7. Tendensen for rammeprogrammerne oligomerise er blevet bredt dokumenteret og kunne i væsentlig grad ændre adfærd (dvs. kinetik, uønskede protein-protein interaktioner og dannelse af aggregater) af kimære proteiner. Brugen af optimerede monomere fluorescerende proteiner bør derfor overvejes 17.
Et andet kritisk aspekt af dynamiske billeddiagnostiske undersøgelser ved hjælp af fluorescens og fotoblegning er den nødvendige tid til at blege fluorescens effektivt og måle fluorescens reselvopdagende (og dermed protein mobilitet) nøjagtigt, hvilket også afhænger af det område af ROI og lokale celle tykkelse. Hvis en given GFP-mærket protein har en høj diffusionskoefficient, kan diffusion ske under blegning og således interferere med genvinding tidsmålinger. For at opnå en hurtig og effektiv blegning, anbefales det kraftigt, at en "zoom ind"-funktionen (hvis den findes), og mere end en laser linje anvendes. Selv om anvendelsen af en hurtig scanning modul (dvs. en resonant scanner) i høj grad kan forbedre hastigheden af billedbehandling i opsvingsfasen af et eksperiment, i vores hænder det også reducerer blegning effektivitet. Alternative systemer til scanning (såsom en roterende skive er udstyret med en dedikeret fotoblegning enhed), og mere kraftfulde lasere kan dog forbedre både blegning effektivitet og erhvervelse hastighed.
De fleste fluorescerende proteiner, der anvendes i FRAP og flip forsøg viser en vis grad af reversibel fotoblegning og blinking der skal overvejes, når du udfører kvantitative analyser. Udsvingene mellem fluorescerende og mørke tilstande forekomme i andet til minut tidshorisont. For EGFP, har det vist sig, at under blegning eksperimenter, kan fluorescens variationer indebære mindre end 10% af molekylerne, og dermed i den nuværende protokol dette fænomen er ubetydelig. Hvis holdes konstant forhold, vil det indføre en konstant skævhed i resultaterne. Hvis der anvendes andre fluorescerende proteiner, hvor den reversible fraktion er betydeligt større (YFP), eller for at påvise og vurdere fotoblegning reversibilitet, kan dette gøres ved at måle fluorescens opsving efter fotoblegning på hele levende celle, hvis der observeres bedring dette kan kun være resultatet af fotoblegning reversibilitet 18.
Den potentielle toksicitet af lys under forsøgene er en anden afgørende faktor, især fordi fotoblegning kræver stærkt lys. Det er well kendt, at ophidset fluorophores en tendens til at reagere med ilt til at producere frie radikaler, der kan påvirke forskellige intracellulære processer og endda celle levedygtighed 19, og så er det nødvendigt at etablere en balance mellem effektiv blegning og minimal fototoksicitet, desuden bør cellelevedygtighed altid kontrolleres efter live-cell imaging eksperimenter. I betragtning af den korte optagetid, vi ikke betragte den genotoksiske effekt af kort bølgelængde lys (405 nm) i det beskrevne i dette papir eksempel, men hvis en længere eksperiment er nødvendig, ikke bør anvendes en 405 nm laser linje.
Vi valgte ikke at bruge en modsvarende tilgang til transmission elektronmikroskopi på grund af den heterogene karakter OSER arkitektur og den kendsgerning, at vi ønskede at observere så mange celler (og strukturer) som muligt. Mangfoldigheden af den fine struktur af protein aggregater i celler kan være et centralt element i forskellige sygdomme, og vi var interesseret i at få en bred vifte af samples, hvorimod en modsvarende tilgang giver mulighed for observation af færre hændelser i samme periode tid. Correlative lys-elektronmikroskopi (CLEM) Imidlertid bør være det første valg, når de undersøger begivenhederne i strukturer, der ikke let kan identificeres (såsom mindre fremtrædende ER underdomæner), eller i et begrænset antal celler (såsom mikro-indsprøjtning celler). Det er værd at bemærke, at vore eksperimenter var kendetegnet ved en høj grad af transfektionseffektivitet (mindst 30% af cellerne blev transficeret), ellers muligheden for at observere Osers strukturer noncorrelatively er ret begrænset.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Forfatterne har ikke noget at afsløre.
Acknowledgments
Forfatterne er taknemmelige for Fondazione Filarete for sin hjælp og støtte i offentliggørelsen af denne artikel. Vi vil også gerne takke Centro Europeo di Nanomedicina for brug af Tecnai G2 transmissions elektron mikroskop.
Materials
| Name | Company | Catalog Number | Comments |
| Dulbecco’s Modified Eagle Medium (DMEM) | Invitrogen | 41966029 | |
| Dulbecco’s Modified Eagle Medium (DMEM) w/o phenol red | Invitrogen | 31053028 | |
| Fetal Bovine Serum (FBS) | Invitrogen | 10270106 | |
| Pen/Strep | Invitrogen | 15140-122 | |
| L-Glutamine 200 mM solution | Invitrogen | 25030-024 | |
| jetPEI | Polyplus Transfection | PP10110 | |
| OxyFluor | Oxyrase Inc. | OF-0005 | |
| Glutaraldehyde Grade I | Sigma Aldrich | G5882 | |
| Sodium Cacodylate Trihydrate | Sigma Aldrich | C0250 | |
| Osmium Tetroxide 4% solution | Electron Microscopy Science | 19150 | |
| Uranyl Acetate Dihydrate | Sigma Aldrich | 73943 | slightly radioactive |
| Propylene Oxide | Sigma-Aldrich | 82320 | |
| EPON embedding medium kit | Sigma-Aldrich | 45359-1EA-F | |
| Lead Citrate | Electron Microscopy Science | 17800 | |
| Bench top centrifuge | Eppendorf | 5415 D | |
| Spectral Confocal Microscope | Leica Microsystems | TCS SP5 | |
| CO2 Microscope Cage Incubation System | OkoLab | ||
| Ultramicrotome | Leica Microsystems | UC6 | |
| Diamond knife | Diatome | Ultra 45 ° | |
| Transmission Electron Microscope | FEI | Tecnai G2 | |
| GraphPad Prism Software | GraphPad Software, Inc | ||
| Steel culture cell chamber for 24 mm coverslip | Bioscience Tools | CSC-25 | |
| Electron Microscopy grids | Electron Microscopy Science | G300Cu |
References
- Fasana, E., et al. A VAPB mutant linked to amyotrophic lateral sclerosis generates a novel form of organized smooth endoplasmic reticulum. FASEB J. 24, 1419-1430 (2010).
- Borgese, N., Francolini, M., Snapp, E. Endoplasmic reticulum architecture: structures in flux. Curr. Opin. Cell Biol. 18, 358-364 (2006).
- Ronchi, P., Colombo, S., Francolini, M., Borgese, N. Transmembrane domain-dependent partitioning of membrane proteins within the endoplasmic reticulum. J. Cell Biol. 181, 105-118 (2008).
- Lippincott-Schwartz, J., Snapp, E., Kenworthy, A.
- Lee, M. C., Miller, E. A., Goldberg, J., Orci, L., Schekman, R. Bi-directional protein transport between the ER and. 20, 87-123 (2004).
- Snapp, E. L., et al. Formation of stacked ER cisternae by low affinity protein interactions. J. Cell Biol. 163, 257-269 (2003).
- Papiani, G., et al. Restructured endoplasmic reticulum generated by mutant amyotrophic lateral sclerosis-linked VAPB is cleared by the proteasome. J. Cell Sci. 125, 3601-3611 (2012).
- Almsherqi, Z. A., Kohlwein, S. D., Deng, Y. Cubic membranes: a legend beyond the Flatland* of cell membrane organization. J. Cell Biol. 173, 839-844 (2006).
- Federovitch, C. M., Ron, D., Hampton, R. Y. The dynamic ER: experimental approaches and current questions. Curr. Opin. Cell Biol. 17, 409-414 (2005).
- Takei, K., Mignery, G. A., Mugnaini, E., Sudhof, T. C., De Camilli, P. Inositol 1,4,5-trisphosphate receptor causes formation of ER cisternal stacks in transfected fibroblasts and in cerebellar Purkinje cells. Neuron. 12, 327-342 (1994).
- Feldman, D., Swarm, R. L., Becker, J. Ultrastructural study of rat liver and liver neoplasms after long-term treatment with phenobarbital. Cancer Res. 41, 2151-2162 (1981).
- Sprocati, T., Ronchi, P., Raimondi, A., Francolini, M., Borgese, N. Dynamic and reversible restructuring of the endoplasmic reticulum induced by PDMP in cultured cells. J. Cell Sci. 119, 3249-3260 (2006).
- Costantini, L. M., Fossati, M., Francolini, M., Snapp, E. L. Assessing the tendency of fluorescent proteins to oligomerize under physiologic conditions. Traffic. 13, 643-649 (2012).
- Pyszniak, A. M., Welder, C. A., Takei, F. Cell surface distribution of high-avidity LFA-1 detected by soluble ICAM-1-coated microspheres. J. Immunol. 152, 5241-5249 (1994).
- Taylor, J. P., Hardy, J., Fischbeck, K. H.
- Winklhofer, K. F., Tatzelt, J., Haass, C. The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 27, 336-349 (2008).
- Borgese, N., Gazzoni, I., Barberi, M., Colombo, S., Pedrazzini, E. Targeting of a tail-anchored protein to endoplasmic reticulum and mitochondrial outer membrane by independent but competing pathways. Mol. Biol. Cell. 12, 2482-2496 (2001).
- Chudakov, D. M., Matz, M. V., Lukyanov, S., Lukyanov, K. A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev. 90, 1103-1163 (2010).
- Bancaud, A., Huet, S., Rabut, G., Ellenberg, J. Fluorescence perturbation techniques to study mobility and molecular dynamics of proteins in live cells FRAP, photoactivation, photoconversion, and FLIP. Cold Spring Harb. Protoc. 2010, (2010).
- Michida, T., et al. Role of endothelin 1 in hemorrhagic shock-induced gastric mucosal injury in rats. Gastroenterology. 106, 988-993 (1994).

{kind=link}