Sequenciação e estudos de genotipagem baseados, incluindo Estudos do Genoma-Wide Association (GWAS), estudos locus candidato, e deep-sequenciação estudos, identificamos muitas variantes genéticas que estão estatisticamente associados com uma doença, característica ou fenótipo. Ao contrário do que as previsões iniciais, a maior parte destas variantes (85-93%) estão localizados em regiões não codificantes e não alteram a sequência de aminoácidos de proteínas de 1,2. Interpretando a função destas variantes não-codificantes e determinar os mecanismos biológicos que ligam-los para a doença associada, traço, ou fenótipo provou desafiador 3-6. Nós desenvolvemos uma estratégia geral para identificar os mecanismos moleculares que ligam variantes para um importante intermediário fenótipo – a expressão do gene. Esse gasoduto é projetado especificamente para identificar a modulação da ligação de variantes genéticas TF. Esta estratégia combina abordagens computacionais e técnicas de biologia molecular voltadas para preverefeitos biológicos das variantes candidatos in silico e verifique estas previsões empiricamente (Figura 1).
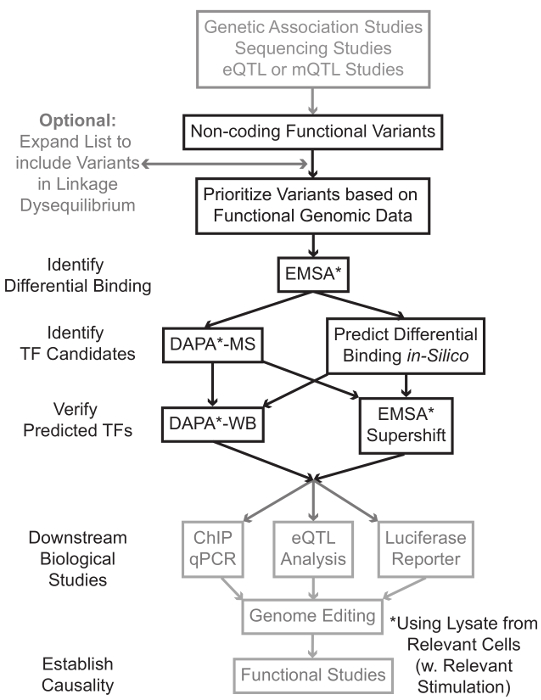
Figura 1:.. Uma abordagem estratégica para a análise de Passos não-codificantes variantes genéticas que não estão incluídos no protocolo detalhado associado a este manuscrito estão com fundo cinza Por favor clique aqui para ver uma versão maior desta figura.
Em muitos casos, é importante começar pela expansão da lista de variantes para incluir todos aqueles em alta linkage-desequilíbrio (LD), com cada variante estatisticamente associada. LD é uma medida da associação não aleatória de alelos em duas posições cromossómica diferente, o que pode ser medido pela estatística de R 2 7. R2 é uma medida da linkage desequilíbrio entre duas variantes, com um r 2 = 1 denotando ligação perfeita entre duas variantes. Os alelos em alta LD são encontrados a co-segregar no cromossoma através de populações ancestrais. matrizes de genotipagem circulante não inclui todas as variantes conhecidas do genoma humano. Em vez disso, eles explorar o LD dentro do genoma humano e incluem um subconjunto das variantes conhecidas que funcionam como proxies para outras variantes dentro de uma região particular de LD 8. Assim, uma variante sem qualquer consequência biológica pode estar associada com uma doença em particular, porque é em LD com a variante a-causal variante com um efeito biológico significativo. Processualmente, recomenda-se para converter a versão mais recente dos 1.000 genomas projetar 9 arquivos de chamada variante (VCF) em arquivos binários compatíveis com Plink 10,11, uma ferramenta de código aberto para análise de associação do genoma inteiro. Posteriormente, todas as outras variantes genéticas com LD r 2> 0,8 com cada va genética de entradaRiant podem ser identificados como candidatos. É importante usar a população de referência apropriado para este passo-por exemplo, se uma variante foi identificada em indivíduos de ascendência europeia, dados de indivíduos de ascendência similar deve ser usado para a expansão LD.
LD expansão muitas vezes resulta em dezenas de variantes de candidatos, e é provável que apenas uma pequena fracção delas contribuir para mecanismo da doença. Muitas vezes, isto não é exequível para examinar experimentalmente cada uma destas variantes individualmente. Portanto, é útil para alavancar os milhares de conjuntos de dados genômicos funcionais publicamente disponíveis como um filtro para priorizar as variantes. Por exemplo, o consórcio CODIFICAR 12 realizou milhares de experiências Chip-seguintes que descrevem a ligação de TF e co-factores, e marcas de histona numa vasta variedade de contextos, juntamente com os dados de acessibilidade cromatina de tecnologias tais como a DNase-SEQ 13, ATAC -seq 14 e FAIRE-seq 15. databases e servidores web, tais como o navegador UCSC Genome 16, Roteiro Epigenomics 17, Blueprint Epigenome 18, Cistrome 19, e remapear 20 permitem o acesso gratuito aos dados produzidos por essas e outras técnicas experimentais através de uma ampla gama de tipos e condições celulares. Quando existem muitas variantes para examinar experimentalmente, estes dados podem ser utilizadas para hierarquizar os localizados dentro das regiões reguladoras prováveis em tipos de células e de tecidos relevantes. Além disso, nos casos em que uma variante está dentro de um pico ChIP-seq para uma proteína específica, esses dados podem fornecer potenciais clientes em potencial quanto à TF específico (s) ou co-fatores cuja ligação pode estar afetando.
Em seguida, as variantes resultantes priorizadas são testadas experimentalmente para validar proteína dependente do genótipo previu ligação usando EMSA 21,22. EMSA mede a mudança na migração do oligo num gel de TBE não redutoras. oligo marcado com fluorescência é incubada com olisado nuclear, e ligação de factores nucleares irá retardar o movimento do oligo no gel. Desta maneira, oligo que se tenha ligado mais factores nucleares vai apresentar-se como um sinal de fluorescência mais forte sobre a digitalização. Notavelmente, a EMSA não requer previsões sobre as proteínas específicas cuja ligação será afetado.
Uma vez que as variantes são identificados que estão localizados dentro das regiões reguladoras previstos e são capazes de factores nucleares de ligação diferencialmente, métodos computacionais são utilizados para prever o TF específico (s) cuja ligação que possa afectar. Nós preferimos usar CIS-BP 23,24, RegulomeDB 25, UNIProbe 26, e Jaspar 27. Uma vez candidato TFs são identificados, essas previsões podem ser testados especificamente utilizando anticorpos contra esses TFs (EMSA-supershifts e DAPA-Westerns). Um EMSA-supershift envolve a adição de um anticorpo específico para o TF ao lisado nuclear e oligo. Um resultado positivo em um EMSA-supershift é represented como mais um desvio na banda de EMSA, ou uma perda da banda (revisto em referência 28). No DAPA complementar, uma cadeia dupla oligo 5'-biotinilado contendo a variante e a 20 pares de bases que flanqueia nucleótidos são incubadas com lisado nuclear do tipo de célula relevante (s) para capturar quaisquer factores nucleares que se ligam especificamente os oligos. O complexo do factor nuclear-duplex oligonucleótido é imobilizado por estreptavidina micropérolas em uma coluna magnética. Os fatores nucleares ligados são recolhidos directamente através de eluição 29,48. previsões de ligação pode então ser avaliada por uma transferência de Western utilizando anticorpos específicos para a proteína. Nos casos em que não existem previsões óbvias, ou muitas previsões, as eluições de variantes menus pendentes dos experimentos DAPA podem ser enviadas para um núcleo de proteômica para identificar TFs candidatos usando espectrometria de massa, que podem posteriormente ser validados usando estes previamente descrito métodos.
No restante do articlE, o protocolo detalhado para análise EMSA e DAPA de variantes genéticas é fornecido.
