Sekventering og genotypebestemmelse baserede undersøgelser, herunder genom-Wide Association Studies (GWAS), kandidat locus undersøgelser, og dyb-sekventering studier har identificeret mange genetiske varianter, der er statistisk forbundet med en sygdom, træk, eller fænotype. I modsætning til tidlige forudsigelser, de fleste af disse varianter (85-93%) er placeret i ikke-kodende regioner og ikke ændrer aminosyresekvensen af proteiner 1,2. Fortolkning af funktionen af disse ikke-kodende varianter og bestemme de biologiske mekanismer, der forbinder dem til den associerede sygdom, er træk, eller fænotype bevist udfordrende 3-6. Vi har udviklet en generel strategi til at identificere de molekylære mekanismer, der knytter varianter til et vigtigt mellemprodukt fænotype – genekspression. Denne rørledning er specielt designet til at identificere modulation af TF binding af genetiske varianter. Denne strategi kombinerer beregningsmæssige metoder og molekylærbiologiske teknikker med henblik at forudsigebiologiske effekter af kandidat varianter i silico og kontrollere disse forudsigelser empirisk (Figur 1).
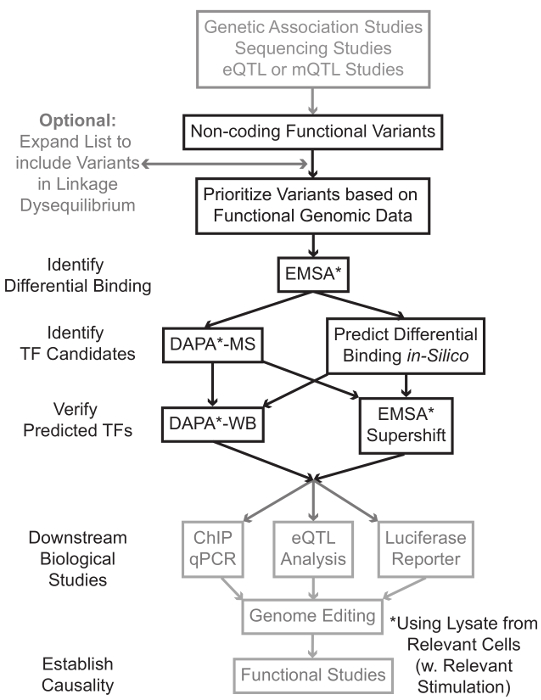
Figur 1:.. Strategisk tilgang til analyse af ikke-kodende genetiske varianter Steps, der ikke er medtaget i detaljeret protokol i forbindelse med dette manuskript er skraveret med gråt Klik her for at se en større version af dette tal.
I mange tilfælde er det vigtigt at begynde med at udvide listen over varianter at omfatte alle dem i high kobling-uligevægt (LD) med hver statistisk associeret variant. LD er et mål for ikke-tilfældige association af alleler ved to forskellige kromosomale positioner, hvilket kan måles ved R2 statistik 7. R2 er et mål for linKage uligevægt mellem to varianter, med en r 2 = 1 angiver perfekt kobling mellem to varianter. Alleler i høj LD viser sig at co-segregerer på kromosomet tværs ancestral populationer. Aktuelle genotype arrays inkluderer ikke alle kendte varianter i det humane genom. I stedet er de udnytte LD inden det humane genom og omfatte en delmængde af de kendte varianter, der fungerer som stedfortrædere for andre varianter inden for en bestemt region i LD 8. Således kan en variant uden biologisk konsekvens være associeret med en bestemt sygdom, fordi det er i LD med den kausale variant-varianten med en meningsfuld biologisk virkning. Proceduremæssigt, anbefales det at konvertere den nyeste version af de 1.000 genomer projekt 9 variant opkald filer (VCF) i binære filer er kompatible med Plink 10,11, et open source-værktøj til hele genomet association analyse. Efterfølgende alle andre genetiske varianter med LD r 2> 0,8 med hver indgang genetiske vaRiant kan identificeres som kandidater. Det er vigtigt at bruge den relevante Referencepopulationen for dette trin for fx hvis en variant blev identificeret i emner af europæisk afstamning, data fra emner af lignende afstamning skal bruges til LD ekspansion.
LD ekspansion resulterer ofte i snesevis af kandidat-varianter, og det er sandsynligt, at kun en lille brøkdel af disse bidrager til sygdom mekanisme. Ofte er det umuligt at eksperimentelt undersøge hver af disse varianter individuelt. Det er derfor nyttigt at udnytte de tusindvis af offentligt tilgængelige funktionelle genomiske datasæt som et filter til at prioritere de varianter. For eksempel har KODE konsortium 12 udført tusindvis af chip-seq eksperimenter beskriver bindingen af TF'er og co-faktorer og histon-mærker i en lang række sammenhænge, sammen med data chromatin accessibility fra teknologier såsom DNase-seq 13, ATAC -seq 14, og FAIRE-seq 15. Databasernes og webservere såsom UCSC Genome Browser 16, køreplanen Epigenomics 17, Blueprint epigenome 18, Cistrome 19, og restere 20 giver fri adgang til data fra disse og andre eksperimentelle teknikker på tværs af en bred vifte af celletyper og betingelser. Når der er for mange varianter at undersøge eksperimentelt, kan disse data anvendes til at prioritere dem, der findes inden for sandsynlige regulatoriske regioner i relevante celle- og vævstyper. I tilfælde, hvor en variant er inden for en chip-seq top for et specifikt protein, kan disse data give potentielle kundeemner som til den specifikke TF (er) eller co-faktorer, hvis binding kan påvirke.
Dernæst resulterende prioriteres varianter screenes eksperimentelt at validere forudsagte genotype-afhængigt protein binding under anvendelse EMSA 21,22. EMSA måler ændringer i vandringen af oligo på et ikke-reducerende TBE gel. Fluorescensmærket oligo inkuberes mednuklear lysat, og binding af nukleare faktorer vil forsinke bevægelsen af oligo på gelen. På denne måde oligo der har bundet flere nukleare faktorer vil præsentere som et stærkere fluorescerende signal ved scanning. Især betyder EMSA ikke kræver forudsigelser om de specifikke proteiner, hvis binding vil blive påvirket.
Når varianter er identificeret som er placeret inden for forudsagte regulatoriske regioner og er i stand til forskelligt bindende nukleare faktorer, der beregningsmetoder ansat til at forudsige den specifikke TF (er), hvis bindende de kan påvirke. Vi foretrækker at bruge CIS-BP 23,24, RegulomeDB 25, UniProbe 26 og Jaspar 27. Når kandidat TF'er er identificeret, kan disse forudsigelser specifikt testet ved hjælp af antistoffer mod disse TF'er (EMSA-supershifts og DAPA-Westerns). En EMSA-supershift indebærer tilsætning af en TF-specifikt antistof til den nukleare lysat og oligo. Et positivt resultat i en EMSA-supershift er Represented som en yderligere forskydning i EMSA band, eller et tab af bandet (revideret i henvisning 28). I den komplementære DAPA, er en 5'-biotinyleret oligo duplex indeholder varianten og 20 basepar flankerende nukleotider inkuberet med nukleare lysat fra relevante celletype (r) til at fange eventuelle nukleare faktorer specifikt binder oligoer. Oligo duplex-nuklear faktor-kompleks immobiliseres ved streptavidin mikroperler i en magnetisk søjle. De bundne nukleare faktorer indsamles direkte gennem eluering 29,48. Bindende forudsigelser kan derefter vurderes ved en Western blot under anvendelse af antistoffer specifikke for proteinet. I tilfælde, hvor der ikke er oplagte forudsigelser, eller for mange forudsigelser, de elueringer fra variant pull-downs af DAPA eksperimenter kan sendes til en proteomics kerne for at identificere kandidat TF'er hjælp massespektrometri, som efterfølgende kan valideres bruge disse tidligere beskrevet metoder.
I den resterende del af article, er den detaljerede protokol for EMSA og DAPA analyse af genetiske varianter forudsat.
