1. Protein Preparation
- DNA cloning.
- Synthesize a DNA sequence of interest, for example, the DNA sequence of NI10C10, or isolate via PCR from the host organism using standard molecular biology techniques11. Flank the gene of interest with restriction sites during synthesis or by placing sites in the 5’- end of the PCR primers to correspond to a module in the plasmid pEMI91 (Addgene #74888)12.
- Separately digest both the plasmid pEMI9112 and the DNA sequence of interest with a pair of restriction sites so that the sequence of interest will be flanked by the tandem I91 repeats (see Figure 1). Follow the standard protocol for the restriction sites.
- Purify the digested product using gel electrophoresis and then ligate the products using T4 DNA ligase, following a standard protocol. After ligation, transform the plasmid into E. coli cells for plasmid purification and purify the plasmid using standard protocols. Sequence using T7 primers or internal pEMI91 primers to verify that the sequence was successfully transformed.
- Transformation.
- Remove protein expression cells, such as C41 (DE3) pLysS cells, from a -80 °C freezer and thaw completely on ice.
- Add 1 µL of plasmid DNA to the cells and stir briefly; pipetting up and down is not advisable as it will introduce air bubbles and warm the cells.
- Incubate the culture tube containing the cells and the plasmid on ice for 30 min.
- Heat shock the cells in a water bath at 42 °C for 45 s.
- Return the tube to ice for 2 min.
- Place 950 µL of LB broth in the cells and shake at 250 rpm for 1 h at 37 °C.
- Place about 200 µL of the transformation onto LB plates containing 100 µg/mL Ampicillin. If using a plasmid other than pEMI91, then use the appropriate antibiotic for that plasmid. Place plate overnight in an incubator at 37 °C. The next day, remove plate from incubator, wrap with parafilm, and store refrigerated up to 1 month.
- Protein expression.
- Inoculate 15 mL of LB broth medium with 100 µg/mL ampicillin in a 50 mL tube at 37 °C for overnight growth by touching a sterile pipette tip to a single bacterial colony on the plate and pipetting up and down in the LB.
- Transfer the 15 mL culture into 1 L of LB broth medium with 100 µg/mL ampicillin and shake for 4-12 h at 37 °C (until OD600 > 0.8).
- Collect 10 mL of the culture for plasmid preparation.
- Add 0.2-1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) and lower the temperature to room temperature for overnight expression.
- Harvest cells the next day by splitting into four tubes and centrifuging at 4,000 × g for 40 min, and then freeze the pellet at -80 °C for several hours.
- Send the plasmids extracted from the cells for sequencing with the primers corresponding to the inserted cassette of the protein12 and verify the fidelity of the inserted DNA.
- Protein purification.
- Thaw one tube of frozen cells for 30 min at room temperature.
- Suspend thawed cells in lysis buffer (38 mL of water, 2 mL of glycerol, 50 mM Tris-HCl pH 7.6, 150 mM NaCl, 1 mM CaCl2, 10 µg/mL DNase, 1 mM PMSF, 1 mM TCEP, 500 µg/mL lysozyme) and shake on ice for 1 h.
- Freeze the lysed cells at -80 °C for several hours and then re-thaw at room temperature.
- Spin the lysate down at 13,100 × g for 30 min at 4 °C.
- Run the supernatant through a gravity flow column for the specific tag (e.g., Strep-tag or His-tag).
- Perform a buffer exchange using a centrifugal filter using the appropriate buffer for Strep-tag or His-tag and store at 4 °C before use.
2. Slides Preparation for Sample Preparation
- Prepare glass slides using Piranha solution.
- Place 10-30 pieces of round glass cover slips (radius of 7.5 mm) into a 40 mL glass beaker.
Note: Perform this step and following steps in a hood. Follow standard procedures for dealing with dangerous chemicals during this step. - Add 30 mL of concentrated (18.4 M) sulfuric acid.
- Add 10 mL of 30% hydrogen peroxide.
- Heat the mixture for 10-30 min to 95 °C.
- Carefully decant the Piranha solution into a separate waste container (to be reused or discarded).
- Rinse the slides with deionized water to remove Piranha solution.
- Suspend the slides in 40 mL of acetone.
- Decant acetone into a waste container and re-suspend slides in 40 mL of ethanol.
- Use clean forceps to carefully extract one slide at a time and dry with argon or purified air.
- Place clean and dried slides under vacuum until the gold evaporation, or under argon for long-term storage.
- Place 10-30 pieces of round glass cover slips (radius of 7.5 mm) into a 40 mL glass beaker.
- Prepare gold coated slides (optional).
Note: This step requires the use of a vacuum (e-beam) evaporator, which is available in many clean room facilities at major universities.- Cut five microscope slides (25 mm x 75 mm rectangular shape) in half using a glasscutter.
- Apply double-sided adhesive tape across the middle of each of the 10 half microscope slides.
- Cut the sticky side of a sticky note and apply to the double-sided tape so that the sticky side of the note is face-up. The sticky note is generally less adhesive, but still firmly attaches to slides to prevent future breaking. Then those half microscope slides can now work as glass slides holder.
- Carefully press four clean glass slides (from section 2.1.10) to each corner of the sticky side of the holder.
- Move the glass slides to an e-beam evaporator. Follow the specifications of the evaporator to apply 70 nm of chromium and 300 nm of gold to the surface.
- Store the gold-coated slides under argon.
3. Sample Preparation
- Prepare slide.
- Select a clean iron disk (15 mm diameter) and attach an adhesive tab to it.
- Unattach the cover piece of the adhesive tab.
- Select a piece of clean glass (from section 2.1.10) or gold (from section 2.2.7) and firmly place it onto the sticky side of the iron disk, this will be the sample slide.
- Deposit protein onto slide.
- Dialyze the polyprotein into the buffer for the experiment (25 mM Tris-HCl, pH 7.6 with 150 mM NaCl) using a buffer exchange column such as a 0.5-mL centrifugal filter. Spin the protein in the column for 10 min at 13,000 rpm and then reverse the column and elute the protein into a new buffer by spinning at 1,000 rpm for 2 min.
- Determine the approximate protein concentration by measuring the absorbance at 280 nm, using a spectrophotometer.
- Dilute the protein to 10-100 µg/mL in a final volume of 100 µL.
- Apply the 60 µL of protein solution onto the center of the slide. Be careful at this stage not to let any liquid enter beneath into the gap between the glass and the iron slide, as this can cause swelling of the adhesive during the experiment and uncontrolled sample movements.
Note: Gold slides are hydrophobic, and a spherical droplet will form, while glass slides are more hydrophilic and the protein solution may spread. - Let the sample sit at room temperature for 10-60 min. During this time, proceed to the next step to begin setting up the atomic force microscope.
4. Atomic Force Microscope (AFM) Setup
Note: The following is a general description for setting up the AFM, and some specific details may differ depending on the specific instrumentation used. The instrumentation used is partially home-built and described in detail in Scholl13.
- Mount the cantilever onto an AFM cell.
- Choose the cantilever with appropriate properties for the application. Use cantilevers with spring constants 4-10 pN/nm for low unfolding forces (~10-50 pN), while use cantilevers with spring constants 15-100 pN/nm for high unfolding forces.
- Carefully pick up the cantilever from the end and place it into the probe holding cell.
- Make sure the holding cell firmly holds the cantilever in place before continuing.
- Place the holding cell into the AFM head for alignment of the laser.
- Align the laser in the AFM head.
- Place the head onto an inverted microscope and connect a battery pack to the AFM head for powering the laser in the head.
- Attach a camera to the microscope detector so that the laser light can be visualized onto a TV or monitor.
- Position the laser so that it is located onto the tip of the cantilever
- Mount head with sample.
- Flush 10 µL of buffer into each of the ports of the probe holding cell.
- Take the sample slide which has been incubating and decant 40 µL of fluid from the incubated solution. Add 40 µL of the buffer on the slide. Place the sample slide onto the magnet above the piezo.
- Make sure that the AFM stage is in the elevated position. Then place the AFM head onto the stage so that the AFM cantilever is above the sample droplet.
- Center the reflected laser beam onto the photodiode.
- Cut a small piece of paper and place it in front of the AFM photodiode.
- Reposition the AFM laser using the knobs so that the laser spot on the paper becomes focused and bright.
- Adjust the AFM head mirror so that the laser hits the photodiode to maximize the total signal in all quadrants (A+B+C+D) and causes the difference signal between top two and bottom two quadrants to be zero (A+B-C-D).
5. Atomic Force Microscope Calibration
- Measure the power spectrum.
- On the filter connected to the AFM, turn the filter setting for the AFM head signal to full bandwidth.
- On the AFM itself, make sure that the piezo is off as this will add noise to the signal.
- Use the AFM software14 to measure the average of 512 calculations of the power spectrum from 1,024 data points per calculation.
- Use the AFM software15 to integrate the power spectral density across the first peak, which corresponds to the main mode of vibration for the cantilever.
- Calculate photodiode sensitivity.
- On the AFM itself, turn the piezo controller on and change the filter setting to 500 Hz (low-pass filter).
- Monitor the difference signal which should be fluctuating around zero in the AFM software. Swiftly move the AFM head down a few hundred micrometers using micropositioners. Repeat moving the head down and monitor the difference signal for consistent jumps. The surface is very nearby when the signal jumps start increasing in height.
- As soon as the deflection signal saturates upon contact with the surface, move the head away from the surface slightly.
- In the AFM software, use the input controls to regulate the piezo voltage to find the surface, by moving up or down 100-5,000 nm. If the surface is still not in reach, continue to reduce the distance between the head and the sample manually.
- In the AFM software, conduct a pulling experiment with a scan size of about 500 nm so that the tip comes in contact for about 100 nm of piezo travel.
- Measure the slope of the linear region of the photodiode signal versus piezo displacement curve where the AFM tip remains in contact with the substrate surface.
- Calculate the spring constant and sensitivity using the slope and the integrated power spectrum (
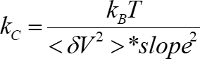 ).
).
6. Data Acquisition
- Preparation.
- On the filters connected to the AFM, set the filter setting so that the sampling frequency is at least twice the bandwidth (Nyquist criterion), which will give an upper bound for the low-pass cutoff.
- Measurements.
- Set the scan size to the total theoretical size of the unfolding polyprotein (number of amino acids × 0.365) plus about 40% to allow for pressing against the substrate. For example, pulling a 9x I91 construct will have a theoretical unfolded length of about 300 nm, so the scan size should be about 420 nm.
- In the AFM software, position the cantilever so that it is 80% of the scan-size away from the surface (in this case, about 340 nm away from the surface).
- In the AFM software, set the scan speed to 300 nm/s initially.
Note: This speed can be modified to be slower, or faster, depending on the application. - Perform a pulling experiment and measure the resulting position of the piezo and the photodiode signal. Use the AFM software to initiate the scan.
- In the AFM software, continue to perform measurements until there are about 10,000 recordings.
Note: Generally, the pickup rate of a positive-controlled molecule is around 0.5%16, so 10,000 are necessary to be able to collect enough data to analyze.
7. Data Analysis
- Normalize data.
- For each recording, compute the extension using extension = displacement – F/kc (kc is from 5.2.7).
- Determine the baseline force level by taking the average of either the beginning or end of the force-extension trace, where no force peaks are present and where the cantilever is not pressing against the surface. The entire force-extension curve can then be shifted by this mean value.
- Translate the data so that the extension is aligned along the zero y-axis. This is because the molecule should be set to zero extension at the beginning of a trace.
- Identifying protein of interest.
- Identify recordings with at least four I91 events that provide a single-molecule fingerprint. These recordings capture true “single-molecule measurements”.
- Save events that are denoted single-molecule events for further analysis.
- Determine contour-length increment.
- For each recording, fit a worm-like chain model to unfolding event,
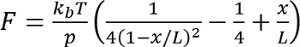 where
where 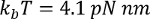 at room temperature, p is the persistence length (typically 0.4 to 1 nm), x is the extension in nanometers, and L is the contour length in nanometers.
at room temperature, p is the persistence length (typically 0.4 to 1 nm), x is the extension in nanometers, and L is the contour length in nanometers.
Note: Form of the worm-like chain is an interpolated solution to the exact, a more exact numerical solution is found in Bouchiat et al.17. Alternate fitting models can be found in Su et al.18. - Calculate the difference between the values for L determined for two consecutive unfolding events, and this difference is coined the “contour-length increment”.
- For each recording, fit a worm-like chain model to unfolding event,
- Determine rupture force.
- Calculate the rupture force for a given unfolding event by taking the highest point before the biggest drop in the unfolding curve.
Representative results from this protocol are shown in Figure 2. Both panels show representative force-extension curves from proteins. The top shows results from a I91 polyprotein, while the bottom shows the I91 protein flanking a protein-of-interest, the NI10C molecule. These recordings show the characteristic force of I91 (200 pN) and contour length increment (28 nm) which indicates that the alignment and calibration of the AFM was successful. These force-extension curves can then be analyzed by worm-like chains (dashed line) which help to determine the force-independent length of the molecule and determine the number of unfolded residues. Once analyzed, the contour-length increments (difference between subsequent contour-lengths) and the unfolding force can be used to determine the protein stability, unfolding rate, and unfolding pathway19.

Figure 1: Plasmid map of polyprotein. This polyprotein plasmid map sequence and physical DNA is available through the Addgene repository (www.addgene.org/74888). It shows the prototypical polyprotein design for AFM, where a polyprotein is composed of 8 identical repeats of I91 protein (gray boxes) which flank a protein of interest (red). Each module contains a unique restriction site which allows customization. Please click here to view a larger version of this figure.
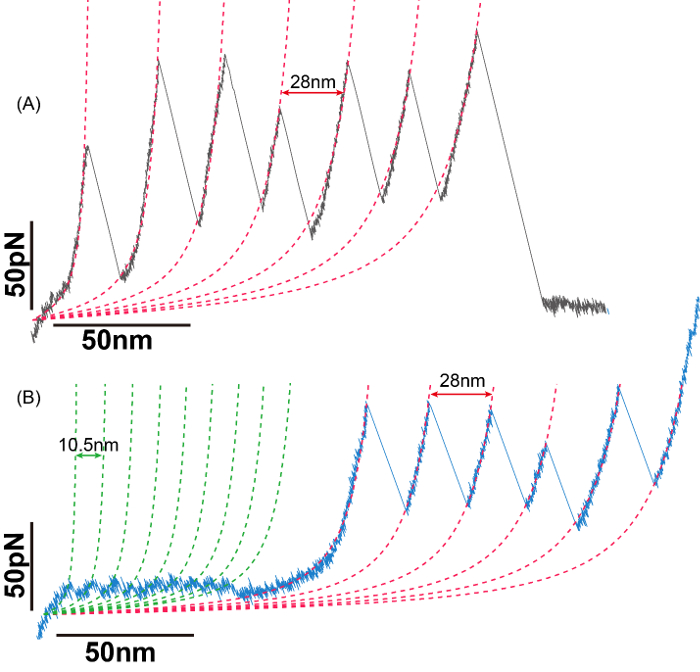
Figure 2: Representative SMFS results. A. Representative force-extension curve for poly I91 protein. Mean contour length increment between peaks is ~28 nm, and unfolding forces are between 100-200 pN. B. Representative force-extension curve for (I91)3-NI10C-(I91)3 protein. Mean contour length increment for ankyrin repeat is ~10.5 nm, and unfolding forces are between 8-25 pN. The regular saw-tooth pattern for ankyrin repeats is followed by I91 unfolding peaks. The pulling speed for both (A) and (B) are 0.02 nm/ms. Please click here to view a larger version of this figure.
