Molti processi biologici verificano presso i siti delle interazioni cellula-cellula, per esempio, cellula-cellula adesione1,2,3, cellula-cellula fusion4 e riconoscimento cellulare5. Tali eventi sono particolarmente importanti durante lo sviluppo degli organismi multicellulari e per la comunicazione cellula-cellula, per esempio, durante le risposte immunitarie. Questi processi sono in genere mediati dalle proteine che sono localizzate alla superficie, cioè, alla membrana del plasma (PM) delle cellule vicine e sottoposti a specifiche interazioni al contatto cellula-cellula che sono precisamente regolamentato in spazio e tempo. In molti casi, queste interazioni sono diretto omo – o eterotipiche interazioni della proteina-proteina trans , ma possono anche coinvolgere gli ioni o ligandi che agiscono come linker extracellulare1. Sebbene di fondamentale importanza, c’è una mancanza di saggi sondando queste interazioni proteina-proteina specifica direttamente nell’ambiente nativo di cellule viventi. Molti metodi di distruzione cellulare (ad es., analisi biochimiche come co-immunoprecipitazione6), richiedono la fissazione (ad es., alcune delle tecniche di Super-risoluzione microscopia ottica e microscopia elettronica delle cellule contatti7), o sono non-specifici, ad esempio, aggregazione / adesione saggi8,9. Per ovviare a questo problema, sono state implementate tecniche di fluorescenza basata sulla fluorescenza resonance energy transfer (FRET)10 o fluorescenza complementazione11. Tuttavia, per raggiungere distanze sufficientemente piccole tra fluorofori, questi metodi richiedono etichette fluorescenti sul lato extracellulare delle proteine10, potenzialmente interferenti con interazioni di trans .
Qui, presentiamo un’analisi di fluorescenza-basata alternativa per le interazioni proteina-proteina a contatti cellula-cellula. Questo approccio combina approcci di cross-correlazione di fluorescenza (scansione spettroscopia di correlazione di fluorescenza (sFCCS), numero di cross-correlazione e luminosità (ccN & B)) e la miscelazione delle cellule che esprimono un costrutto di fusione della proteina di interesse, ad esempio, un recettore di adesione. I recettori studiati nelle due cellule interagenti sono etichettati con due proteine fluorescenti spettralmente separate (FPs), dall’intracellulare (Vedi Figura 1A).
I metodi impiegati sono basati sull’analisi statistica delle fluttuazioni di fluorescenza indotta dal movimento diffusivo delle proteine di fusione fluorescenti attraverso il volume focale di un microscopio a scansione confocale del laser. Più in dettaglio, il test sonde co-diffusione delle proteine di interesse in entrambi vicini PMs a contatti cellula-cellula. Se le proteine subiscono interazioni trans , questi complessi trans porterà proteine fluorescenti che emettono in entrambi i canali spettrali, causando fluttuazioni di fluorescenza correlata di entrambi gli emettitori. D’altra parte, se si verifica alcuna associazione, le fluttuazioni numero di proteine nell’affrontare PMs sarà indipendente, non causando fluttuazioni correlate. L’acquisizione può essere eseguita in due modi: 1) sFCCS si basa su una scansione a forma di linea attraverso il contatto cellula-cellula e sonde in modo efficace le interazioni in un punto situato nella regione del Contatta. Attraverso un’analisi temporale delle fluttuazioni di fluorescenza, sFCCS fornisce anche informazioni dinamiche, cioè, i coefficenti di diffusione dei complessi della proteina; 2) ccN & B si basa su un’analisi di immagine di una sequenza di immagini acquisite presso le regioni di contatto cellula-cellula. Esso ha la capacità di sondare e mappa interazioni lungo tutta la regione (in un piano focale) di contatto, ma non fornisce informazioni sulle dinamiche. Entrambi i metodi possono essere combinati con un’analisi della luminosità molecolare, cioè, il segnale di fluorescenza media emessa nell’unità di tempo da complessi proteici diffusore singolo e, quindi, fornire stime della stechiometria dei complessi della proteina a contatti cellula-cellula.
In questo articolo, forniamo dettagliati protocolli per la preparazione del campione, calibrazione dello strumento, acquisizione dati e analisi ad eseguire l’analisi presentata su un commerciale scansione microscopio confocale del laser. Gli esperimenti possono essere eseguiti su qualsiasi strumento dotato di conteggio di fotoni o rivelatori analogici e un obiettivo con apertura numerica elevata. Abbiamo ulteriormente discutere passaggi critici del protocollo e forniscono schemi di correzione per diversi processi che causano fluttuazioni artefactual segnale, ad esempio, rivelatore rumore, movimento photobleaching o cella. Originariamente sviluppato per sondare le interazioni tra cellule aderenti, il dosaggio può essere modificato per cellule in sospensione, o adattati ai sistemi di membrane modello, ad esempio, le vescicole unilamellari gigante (GUV) o plasma gigante vescicole di membrana (GPMVs), che consente la quantificazione delle interazioni in lipidi diversi ambienti o in assenza di un citoscheletro organizzato12,13.
Spettroscopia di correlazione di fluorescenza di scansione è una versione modificata di spettroscopia di correlazione di fluorescenza14 ed è stato specificamente progettato per sonda dinamica diffusiva lenta del lipido membrane15. Si basa su un’acquisizione di scansione linea perpendicolare al PM contenenti le proteine fluorescenti di interesse. Per sondare le interazioni delle due specie di proteina marcata in modo diverso, l’acquisizione viene eseguita in due canali spettrali utilizzando due linee laser e due finestre di rilevamento per fluorofori spettralmente separati. A causa della dinamica lenta diffusione di proteine nel PM (D≤ ~ 1 µm2/s), una misura di cross-talk-free possa essere eseguita alternando lo schema di eccitazione da linea a linea15. L’analisi inizia con: 1) un algoritmo di allineamento correggere per movimento laterale cellulare basato su block-wise con una media di ~ 1000 righe, 2) determinazione della posizione con fluorescenza massimo segnale, cioè, il PM posizione ogni blocco e 3) mutevole di tutti i blocchi per una comune origine12,15, separatamente in ciascun canale. Quindi, una selezione automatica dei pixel corrispondenti al PM viene eseguita selezionando la regione centrale da una vestibilità gaussiana della somma di tutte le linee allineate (cioè, centro ± 2.5σ). Integrazione del segnale in ogni linea produce la serie temporale fluorescenza a membrana f (t) in ciascun canale (g = verde canale, r = canale rosso). Nota che la dimensione in pixel deve essere abbastanza piccola, ad esempio, < 200 nm, per ricostruire la forma del punto di diffondere funzione e trovare il suo centro, corrispondente alla posizione del PM. In presenza di sostanziale photobleaching, le serie temporali di fluorescenza in ciascun canale possono essere modellate con una funzione di doppio-esponenziale e poi corretto con la seguente formula:16
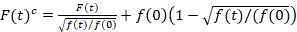 . (1)
. (1)
È importante notare che questa formula corregge efficacemente sia le ampiezze e tempi di diffusione ottenuti da analisi di correlazione di f (t)c, rispetto alle stime dei parametri che si otterrebbe dalla non corretta f (t). Quindi, le funzioni di auto – e cross-correlazione (ACFs / CCFs) della fluorescenza segnali vengono calcolati:
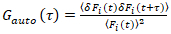 , (2).
, (2).
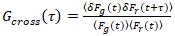 , (3).
, (3).
dove δFho = Fio(t) – 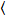 Fio(t)
Fio(t)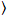 ed io = g, r.
ed io = g, r.
Un modello bidimensionale diffusione è poi montato su tutte le funzioni di correlazione (CFs):
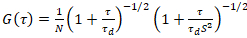 . (4)
. (4)
Qui, N indica il numero di proteine fluorescenti nel volume di osservazione e τd il tempo di diffusione per ogni canale. Questo modello prende in considerazione che in ambito sperimentale descritto, diffusione delle proteine nel PM si verifica nel piano x-z, in contrasto con la configurazione comunemente usata di correlazione di fluorescenza spettroscopia (FCS) esperimenti sulle membrane di sondaggio diffusione nel piano x-y del confocale volume17. La vita w0 e il fattore di struttura S, descrivendo l’allungamento wz del volume focale in z, S = wz/w0, sono ottenuti da una misura di calibrazione punto FCS effettuata con coloranti spettralmente simili e stesse impostazioni ottiche utilizzando valori già disponibili per il coefficente di diffusione Dtintura:
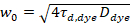 , (5).
, (5).
dove τd, colorante è il tempo di diffusione medio misurato delle molecole della tintura, ottenuta da un modello tridimensionale diffusione ai dati, di montaggio tenendo le transizioni di conto di una frazione T di tutte le molecole di N per un stato di tripletto con una costante di tempo ττ:
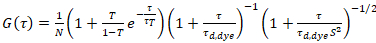 . (6)
. (6)
Infine, i coefficenti di diffusione (D), i valori di luminosità molecolare (ε) e la relativa correlazione incrociata dei dati sFCCS (rel.cc.) sono calcolati come segue:
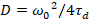 , (7).
, (7).
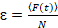 , (8).
, (8).
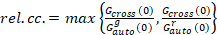 , (9).
, (9).
dove Gcross(0) è l’ampiezza della funzione di correlazione incrociata e 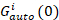 è l’ampiezza della funzione di autocorrelazione nel canale dei -esima.
è l’ampiezza della funzione di autocorrelazione nel canale dei -esima.
Questa definizione della relativa correlazione incrociata, cioè utilizzando max invece di dire all’equazione 9, tiene conto del fatto che il numero massimo dei complessi di due specie proteiche presenti alle concentrazioni differenti è limitato per il specie presenti in un numero inferiore.
Luminosità e il numero di Cross-correlazione si basa su un’analisi del momento dell’intensità di fluorescenza per ogni pixel di una serie di immagini acquisite nel tempo ad un in posizione fisso nel campione, costituiti in genere da ~ 100-200 fotogrammi, con due spettrali canali ( g = verde canale, r = canale rosso). Dalla media temporale hoe la varianza 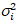 , la luminosità molecolare εi e numero nmi vengono calcolate in ogni pixel e canale spettrale (io = g, r)18:
, la luminosità molecolare εi e numero nmi vengono calcolate in ogni pixel e canale spettrale (io = g, r)18:
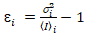 , (10).
, (10).
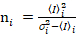 . (11)
. (11)
È importante notare che le equazioni determinate si applicano al caso ideale di un vero rilevatore photon-counting. Per sistemi di rivelazione analogica, le seguenti equazioni applicano19,20:
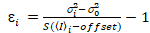 , (12).
, (12).
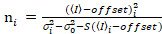 . (13)
. (13)
Qui, S è il fattore di conversione tra fotoni rilevati ed i conteggi digitali registrati, 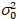 è il rumore di lettura e offset si riferisce all’offset di intensità del rivelatore. Generalmente, queste quantità devono essere calibrate, per qualsiasi tipo di rivelatore, basato su misura la varianza del rivelatore in funzione dell’intensità per illuminazione costante19, ad esempio, una superficie di metallo riflettente o soluzione colorante secchi. L’ offset può essere determinato misurando la velocità di conteggio per un campione senza luce di eccitazione. Eseguendo una regressione lineare della varianza rivelatore-associated
è il rumore di lettura e offset si riferisce all’offset di intensità del rivelatore. Generalmente, queste quantità devono essere calibrate, per qualsiasi tipo di rivelatore, basato su misura la varianza del rivelatore in funzione dell’intensità per illuminazione costante19, ad esempio, una superficie di metallo riflettente o soluzione colorante secchi. L’ offset può essere determinato misurando la velocità di conteggio per un campione senza luce di eccitazione. Eseguendo una regressione lineare della varianza rivelatore-associated 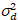 versus trama di intensità (mi), S e può essere determinato19:
versus trama di intensità (mi), S e può essere determinato19:
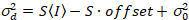 . (14)
. (14)
Infine, la luminosità di cross-correlazione è calcolata in ogni pixel ed è definita in generale come21
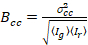 , (15).
, (15).
dove 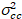 è la croce-varianza
è la croce-varianza 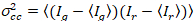 .
.
Per filtrare le fluttuazioni longeve, tutti ccN & calcoli B vengono eseguiti seguendo un vagone coperto del filtro, in modo indipendente per ogni pixel22. Brevemente, ni, εho (ho = g, r) e Bcc sono calcolati in segmenti di ad esempio, 8-15 infissi scorrevoli. I valori così ottenuti possono essere fatte la media poi per ottenere il pixel finale numero e la luminosità.
Analisi di stechiometria
Al fine di stimare la stechiometria dei complessi della proteina a contatti cellula-cellula, la luminosità molecolare possa essere analizzata separatamente in ciascun canale spettrale per il sFCCS o ccN & dati B. In sFCCS, si ottiene un valore di luminosità per misura in ciascun canale. In ccN & B, si ottiene un istogramma di luminosità di tutti i pixel corrispondenti al contatto cellula-cellula e il valore medio (o mediano) può essere utilizzato come luminosità rappresentativo per la misurazione. Eseguendo la stessa analisi su un riferimento monomerico, tutti i valori di luminosità possono essere normalizzati per ottenere direttamente lo stato oligomerico medio dei complessi proteici rilevati. A questo punto, è importante correggere per la presenza di FPs non fluorescente che può comportare una sottovalutazione dello stato oligomerico. Questo viene in genere eseguito misurando la luminosità di un homo-dimerica riferimento proteina23,24 utilizzando SFC un colore o numero e luminosità (N & B).
