The schematic representation of the FbioCLIP-seq procedure is shown in Figure 1. Compared with FLAG-mediated or streptavidin-mediated one-step affinity purification, FLAG-biotin tandem purification removed almost all the copurified proteins, avoiding the contamination of indirect protein-RNA interactions (Figure 2). Representative results for FbioCLIP-seq for LIN28 and WDR43 are depicted in Figure 3 and Figure 4. We performed LIN28 or WDR43 FbioCLIP-seq with mESCs. Figure 3A shows the track view of LIN28 and WDR43 FbioCLIP-seq in pre-let-7g. The reported GGAG motif in pre-let-7g and the cross-linked sites identified by FbioCLIP-seq are shown in Figure 3B. The classification of the mutation sites called by CIMS algorithm showed that LIN28 prefers to bind and cross-link to G nucleotide (Figure 3C). The enriched RNA motifs in LIN28 FbioCLIP-seq binding sites are shown in Figure 3D. More representative tracks of FbioCLIP-seq on LIN28 and HNRNPU are shown in Figure 3E. Comparison of WDR43 and LIN28 FbioCLIP-seq showed their different binding patterns in Rn45 pre-rRNA locus (Figure 4). Figure 5 shows the representative results of FLAG and biotin tag validation after cell line construction. Figure 6 shows representative results of efficient (Figure 6A) or unsuccessful (Figure 6B) FLAG-IP and FLAG-biotin tandem purification.
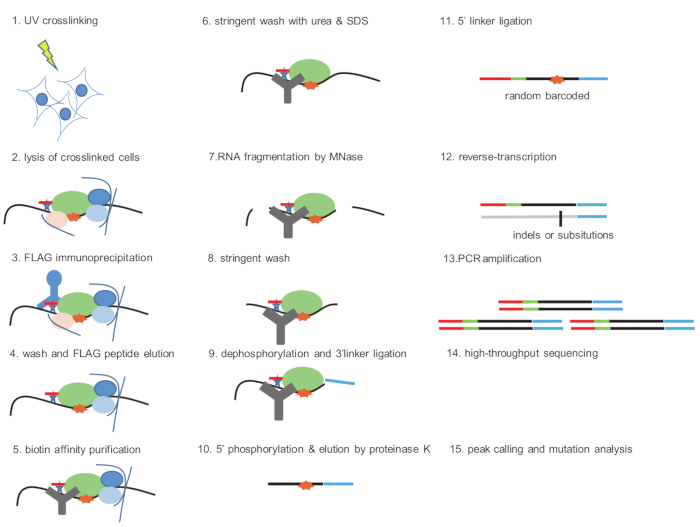
Figure 1: Schematic representation of FbioCLIP. UV-cross-linked cells (step 1) are lysed in lysis buffer (step 2). The FBRBP-RNA complex is immunoprecipitated using anti-FLAG resin (steps 3−4). The eluted protein-RNA complex is further purified with streptavidin beads and stringent wash conditions are used to remove protein-protein interactions (steps 5−6). RNAs are partially digested by MNase and non-protein-bound RNA fragments are removed by further wash (steps 7−8). The RNAs are then dephosphorylated and ligated with 3’ linker (step 9). Then the RNAs are phosphorylated and eluted by proteinase K treatment (step 10). Purified RNAs are ligated with a 5’ RNA linker containing a 6 nt random barcode (step 11). After reverse transcription (step 12), the cDNA library is amplified with PCR and high-throughput sequencing is performed (steps 13−15). Please click here to view a larger version of this figure.
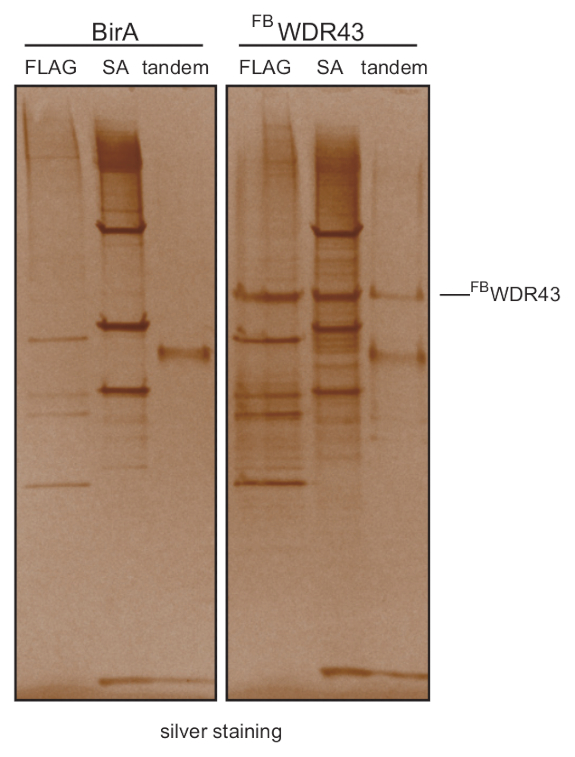
Figure 2: FLAG-biotin tandem purification removes copurified proteins. Silver staining of FBWDR43 showed that almost all the interacting proteins presented in FLAG- or biotin-mediated one-step purification were eliminated after stringent wash in tandem purification. FLAG: FLAG-mediated affinity purification, SA: streptavidin-mediated biotin purification, tandem: FLAG-biotin tandem purification. Please click here to view a larger version of this figure.
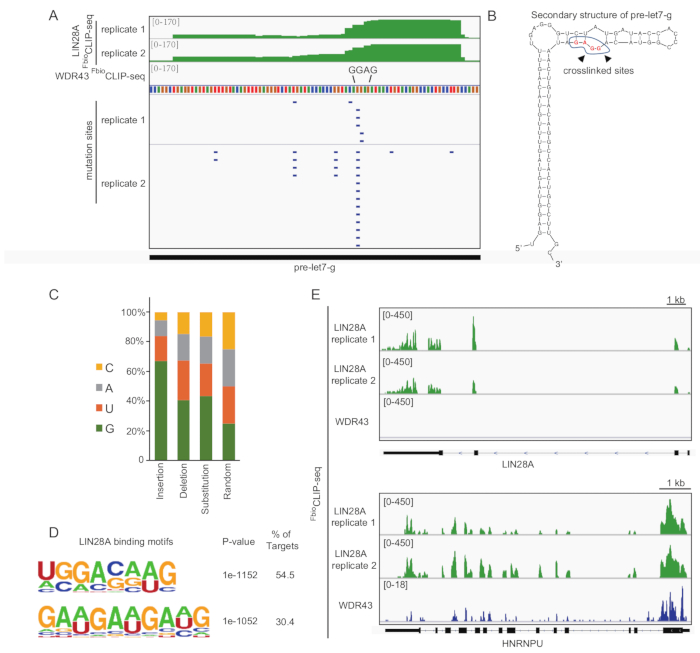
Figure 3: Representative results of LIN28 FbioCLIP-seq. (A) Track view of LIN28 FbioCLIP-seq tags and mutation reads called by CIMS in pre-let-7g RNA. The tags shown are unique reads of FbioCLIP-seq. In total, ~7.7 million unique reads for LIN28 FbioCLIP-seq and ~2.2 million unique reads for WDR43 FbioCLIP-seq were retrieved after removing redundant reads. (B) Cross-linked sites on the GGAG motif of pre-let7-g RNA by LIN28 FbioCLIP-seq. The arrows indicate the cross-linked sites. (C) Percentage of mutated nucleotides in different types of mutations. G is the most frequent cross-linked and mutated nucleotide. Random: random distribution of the four nucleotides. (D) Predicted LIN28 binding motifs by FbioCLIP-seq with HOMER algorithm. (E) Track views of LIN28 FbioCLIP-seq on LIN28 and HNRNPU mRNAs. Please click here to view a larger version of this figure.
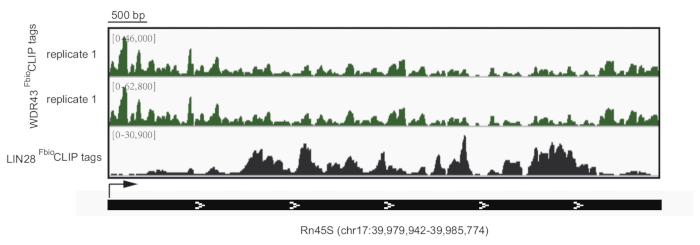
Figure 4: Representative results of WDR43 and LIN28 FbioCLIP-seq on Rn45S locus. Track view of WDR43 and LIN28 FbioCLIP-seq on Rn45S locus. WDR43 and LIN28 showed different binding patterns on pre-rRNA. Please click here to view a larger version of this figure.
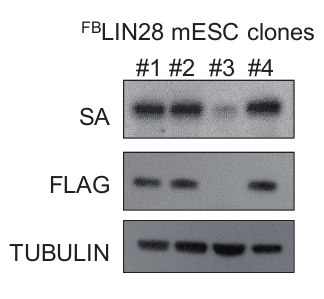
Figure 5: Representative results of FLAG and biotin tag validation. FLAG or biotin Western blot validates the tagging of the cell lines. Clones #1, #2, and #4 showed efficient expression and biotinylation of the tag while #3 showed poor expression of the tagged protein. Please click here to view a larger version of this figure.
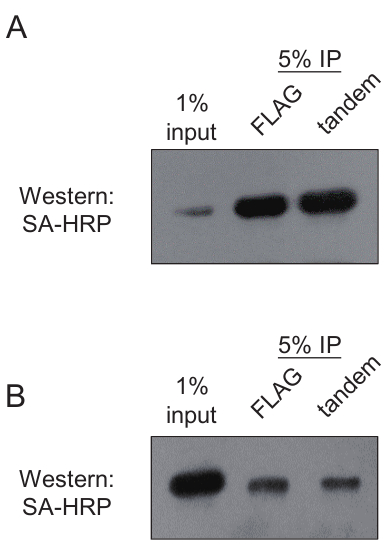
Figure 6: Representative results of FLAG immunoprecipitation and tandem purification efficiency validation. (A) Example of efficient purification of tagged protein by FLAG and biotin affinity purification. (B) Example of inefficient purification of tagged protein by FLAG and biotin affinity purification. Please click here to view a larger version of this figure.
| Buffer | Composition |
| SDS loading buffer | 50 mM Tris pH 6.8, 2% SDS, 0.1% bromophenolblue, 10% glycerol, 100 mM DTT |
| Wash buffer A | 1x PBS, 0.5% NP-40, 0.5% sodium deoxycholate, 0.1% SDS |
| Wash buffer B | 5x PBS, 0.5% NP-40, 0.5% sodium deoxycholate, 0.1% SDS |
| Wash buffer C | 50 mM Tris pH 7.4, 2% SDS |
| Wash buffer D | 5x PBS, 0.5% NP-40, 0.5% SDS, 1 M urea |
| PNK buffer | 50 mM Tris pH 7.4, 0.5% NP-40, 10 mM MgCl2 |
| MNase reaction buffer | 10 mM Tris pH 8.0, 1 mM CaCl2 |
| PNK+EGTA buffer | 50 mM Tris pH 7.4, 0.5% NP-40, 10 mM EGTA |
| Proteinase K digestion buffer | 50 mM Tris pH 7.4, 10 mM EDTA, 50 mM NaCl, 0.5% SDS, 20 µg of proteinase K |
Table 1: Composition of buffers used in this study.
| Oligo name | Sequence | Notes | ||||
| 3' linker | rAppAGATCGGAAGAGCACACGTCT-NH2 | |||||
| 5' RNA linker | GUUCAGAGUUCUACAGUCCGACGUCNNNNN | |||||
| RT primer | AGACGTGTGCTCTTCCGATCT | |||||
| Forward primer 1 | GTTCAGAGTTCTACAGTCCGACGATC | |||||
| Reverse primer 1 | AGACGTGTGCTCTTCCGATCT | |||||
| Forward primer 2 | AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGAC | |||||
| Reverse primer 2 | CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC-s-T | The red and underlined sequences represent Illumina index sequence. | ||||
Table 2: Oligonucleotides used in this study.
