The software analyzes bright field domain images that are off-white and black. The Escherichia coli will look like black oblong shapes on an off-white background and dynamic range of luminance should show a spike at its center (Figure 1). In fluorescent images cells may have a small halo but individual cells with oblong shapes can still be resolved. A mitosis event should be first detected after 30 minutes. Microscope focus should remain stable over time and although cells might move slowly during those 30 minutes, they are unlikely to leave the field of view. Such an experiment will be considered good and can be viewed at Figure 2. Cells might be hard to detect in the bright field domain at low magnification. We recommend establishing the average focus distance with a high copy number (HCN) plasmid, as it is easy to notice at low magnification. Set the average distance while measuring a low copy number (LCN) plasmid at the high magnification in advance. This focus distance depends mainly in the plates, the microscopy system and oil, and not the thickness of gel or Escherichia coli strain.
When adapting this protocol to other labs, the following issues may arise (1) Escherichia coli plates (samples) prepared at an incorrect dilution ratio can show unchanging numbers of cells (Figure 3 and Figure 4) (2) Samples prepared without sealing the plate can show excessive shrinkage. This can be observed as cells drift (Figure 3) or cause a loss of focus (Figure 5) (3) Some samples may exhibit slow shifting 'fixed' cells (in black) on a background of swimming cells (in white). This is likely due to a wet sample and it usually stabilizes as the excess water is absorbed by the gel (Supplementary Video 1) (4) Samples that exhibit no cells after a few minutes of warming up might have been prepared incorrectly, likely due to: (I) Gel was poured while still too hot, (II) protective cap was forgotten, (III) minimal media lacks an ingredient, (IV) incorrect cell density, and (V) gel was sealed too tightly. It is important to punch holes to allow gas exchange at the expense of gel shrinkage (Figure 5) and (VI) cells can also indent the gel in search for nutrients, stacking one on top of the other, compromising the cell monolayer effectively preventing detection of cells and measuring of reliable fluorescent signal (Supplementary Figure 5).
We have measured three circuits in this work. First, a constitutive promotor that regulates GFP shown in Figure 6. Second, a constitutive promotor that regulates super-fold GFP31 (sfGFP) shown in Figure 7. An ssrA degradation tag (AAV) was added to sfGFP to reduce itshalf-life to minutes5. The third circuit is based on a positive feedback circuit1, and is induced by acyl-homoserine-lactone (AHL). The PluxR promoter regulates sfGFP and LuxR, a transcription factor binding with AHL to activate PluxR as shown in Figure 8. Figure 9 presents the dynamics of cell growth (cell numbers versus time), Figure 10 shows the dynamics of the measured signal (mean fluorescence versus time), and Figure 11 shows dynamics of evaluated total noise (standard deviation (STD) versus time).
The SNR (or CV) is widely used in designing analog electronic circuits to express the precision and reliability of the circuit32. CV relates to the distribution of signal between single cells and allows comparison across methods and different equipment such as microscopes and flow analyzers. Calculating SNR from microscope images allows us to compare circuits across time, segmented cells are measured at the same time as providing a measure for the specific resolution of the noise compared to the signal, or a noise interval for a specific time and inducer concentration. This may indicate if detector cells will be able to resolve the exact signal response to the inducer concentration. In this work, the CV was calculated by considering all segmented artifacts which are cells, regardless of the cell progression in the division cycle. SNR was calculated for specific cell area range over time, and then it was averaged for three repeated experiments. Neither the acquired signal nor the STD are reliable alone, as they are specific to the experiment and equipment used. The signal is measured with arbitrary units which depends on equipment gain preset, photo-detector and exposure time. The data presented in Figure 10 suggest that the cells stage in the division cycle does not affect the noise level, as different ranges of area (points in the division cycle) show the same trend. This observation might support the claim that tracking exact mother – daughter relations can be avoided for measuring noise and this could improve the SNR for the presented method. No substantial change in SNR was observed between GFP to sfGFP as shown in Figure 12. We calculate the SNR (SNR= mean/ STD) and present it in Figure 12 and Figure 13.
We then calculated the variance and CV based on the following equations31
1.1 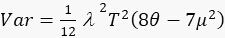
1.2 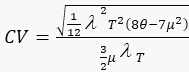
Where λ is protein folding time, T is cell division time, θ and µ are the number of plasmids before and after division, the plasmid copy number31. Using the above equations (1.1, and 1.2) we can fit the data from the flow analyzer signal for gamma distribution.
Then we compared between the CV, which is measured and calculated by flow analyzer (Figure 14), and by the protocol (Figure 15).

Figure 1: Bright field exposure image (a) Stabilized acquisition in bright field (b) Segmentation image, only colored cells enter the calculations (c) Pixel brightness map of the acquisition, spike is the background noise. Thresholding by it segments only Escherichia coli. Please click here to view a larger version of this figure.
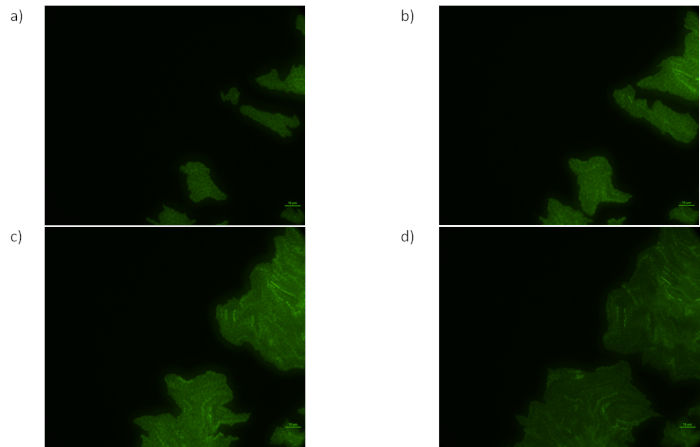
Figure 2: This experiment shows good healthy cells dividing and reacting after 120 minutes. Images were acquired successively at 20 minute intervals. Colonies are in focus, gel is stable between samplings. Colonies divide and produce strong signals. Please click here to view a larger version of this figure.

Figure 3: Effects of incorrect dilution ratio and gel shrinkage on fluorescent image. Images were acquired at 20 minute intervals (a) Individual Escherichia coli circled in red (b) The same cell drifts to the right of the field of view (c) The cell drifts further. The cells also do not divide or change positions, probably due to a low dilution ratio and gel shrinkage. Please click here to view a larger version of this figure.
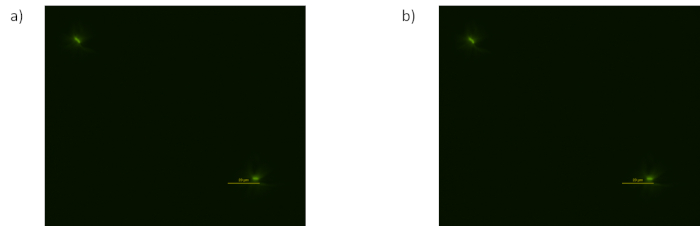
Figure 4: In this experiment no activity was detected and almost no cells were present. Possible reasons are the gel being too hot, the drying of the sample being too aggressive or having no protective cap (a) Fluorescent images taken at 40 minutes (b) Fluorescent images taken at 60 minutes. Please click here to view a larger version of this figure.
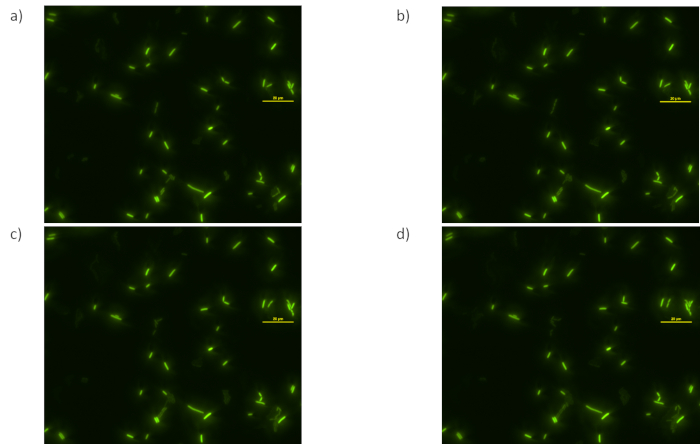
Figure 5: Focus loss and photobleaching.Successive images of (a) through (d) were acquired at 15 minute time intervals using auto focus system. See Supplementary Video 2. Please click here to view a larger version of this figure.
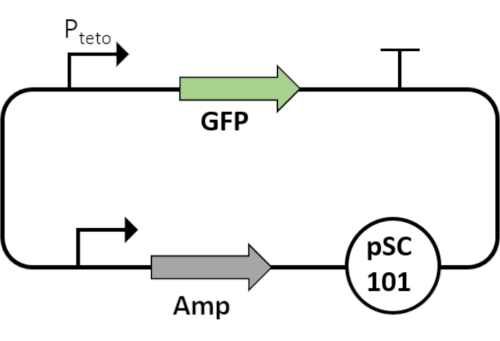
Figure 6: Plasmid map of Pteto that regulates GFP. Without the TetR repressor, the PtetO promoter acts as a constitutive promoter. The circuit is cloned on low copy number plasmids. This plasmid acts as a basis for SNR measurement for any modified circuit. Please click here to view a larger version of this figure.
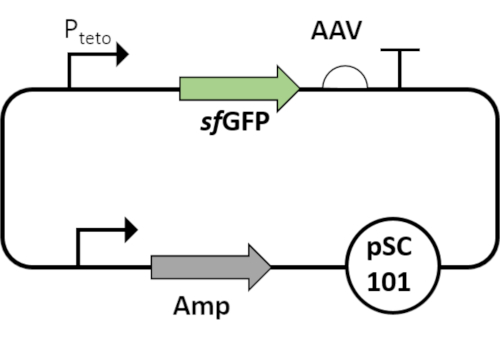
Figure 7: Plasmid map of Pteto that regulates sfGFP. The sfGFP is fused to an AAV degradation tag. The circuit is cloned on low copy number plasmids. The sfGFP-AAV is a more robust variant of GFP, while the AAV tag rendering it susceptible to degradation by housekeeping proteases of the Escherichia coli. Please click here to view a larger version of this figure.
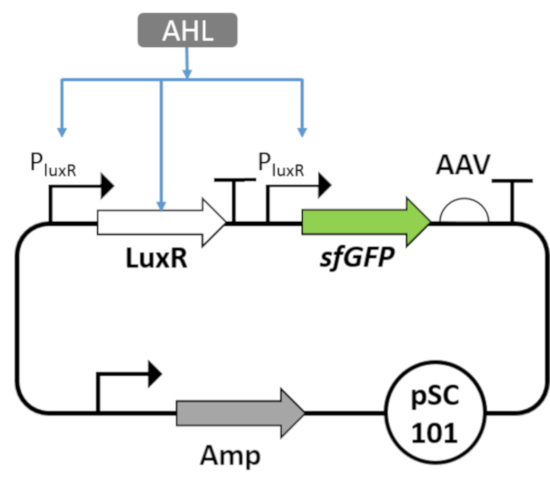
Figure 8: Plasmid map of positive feedback circuit. The circuit is induced by an AHL inducer, which binds LuxR transcription factor. The PluxR promoter, which is regulated by AHL-LuxR complex, actives the production of LuxR and sfGFP-AAV. The circuit is cloned on low copy number plasmids. Please click here to view a larger version of this figure.
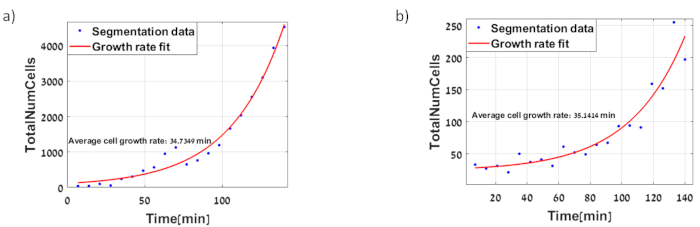
Figure 9: Dynamics of Escherichia coli MG1655 strain growth in minimal media includes (a) PtetO-GFP, exponential growth of about 35 minutes. The images of this experiment are shown in Figure 2 (b) PtetO-sfGFP, exponential growth of about 35 minutes. Please click here to view a larger version of this figure.
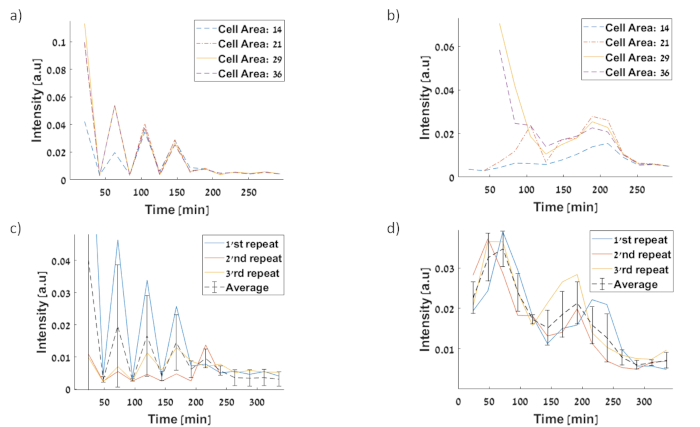
Figure 10: Fluorescent intensity level, normalized per pixel and acquired at 20 minute intervals.
Cells are binned according to area, in order to minimize artifacts. Cells are divided by their area in a micrometer square (a) Pteto-GFP circuitfirst repetition for intensity at 210 minutes is 0.004 a.u (b) PtetO–sfGFP circuit first repetition for intensity at 210 minutes is 0.022 a.u (c) Measured signal of Pteto-GFP circuit (d) Measured signal of PtetO–sfGFP circuit. All experimental data represents the average of three experiments. The software can sort cell area to different groups (a) and (b) show graphs for four cell area ranges representing the division cycle. First area of 14 micrometers represents cells after division as most likely that cells after division will be the smallest. The second area of 21 µm represents cells before division. Third and fourth areas represents cells at division as the total area is multiplied by two (29 and 36 µm) in order to consider cells that took longer to divide. The areas were chosen by manually assessing the data looking on microscopy images. Please click here to view a larger version of this figure.
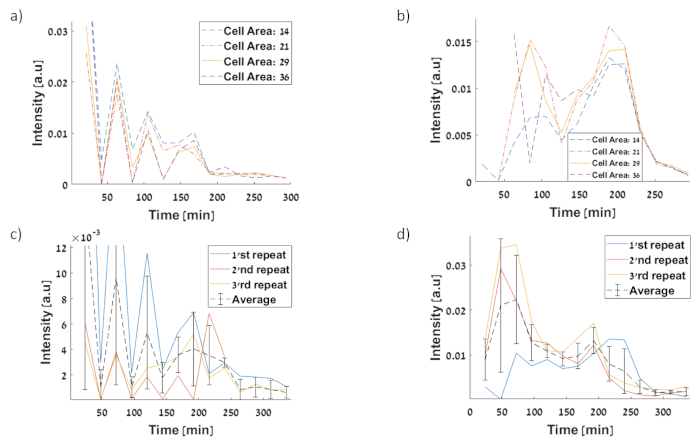
Figure 11:Standard deviation (STD) of single cell fluorescence intensities for: (a) PtetO-GFP circuit first repetition for STD at 210 minutes is 0.001865 a.u (b) PtetO–sfGFP circuit first repetition for STD at 210 minutes is 0.01477 a.u (c) STD of Pteto-GFP circuit (d) STD of PtetO–sfGFP circuit. Please click here to view a larger version of this figure.
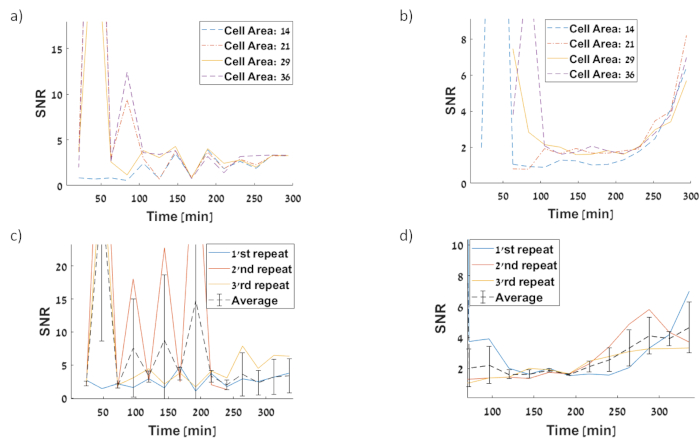
Figure 12:SNR of single cell fluorescence intensities. We calculate the SNR=Mean/STD (a) PtetO-GFP circuit first repetition for SNR at 210 minutes is 1.309 a.u (b) PtetO–sfGFP circuit first repetition for SNR at 210 minutes is 1.29 a.u (c) Three repetitions of GFP measurement (d) Three repetitions of sfGFP measurement. Please click here to view a larger version of this figure.
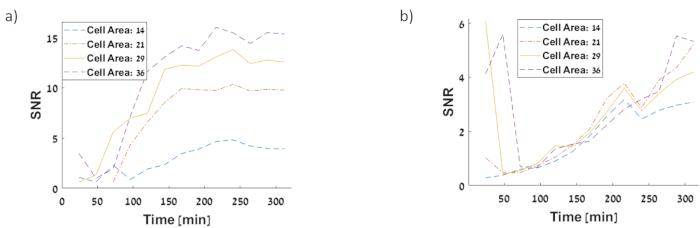
Figure 13:SNR of single cell fluorescence intensities (a)PluxR–sfGFP circuit SNR for saturation response to AHL (b) PluxR–sfGFP circuit SNR for half time response to AHL. The SNR of positive feedback AHL regulated circuit is higher than SNR for constitutive sfGFP circuit. Please click here to view a larger version of this figure.
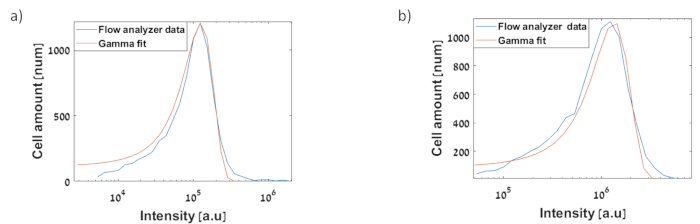
Figure 14: Histograms of the gene circuits based on the flow analyzer: experimental data (blue) and stochastic model data (red). The x axis represents arbitrary fluorescence units from flow cytometry, and the y axis represents the frequency of cells producing the corresponding fluorescence level. The data was measured using a flow analyzer after three hours. GFP fluorescence was quantified by excitation at a wavelength of 484 nm and emission at a wavelength of 510 nm. PE-TexasRed filter voltages were used on a high throughput sampler to measure GFP expression levels. The flow analyzer voltages were adjusted using software so that the maximum and minimum expression levels could be measured with the same voltage settings. Thus, consistent voltages were used across each entire experiment. The same voltages were used for subsequent repetitions of the same experiment. Fitting was made by use of gamma distribution31. This method assumes that the signal depends mostly on random distribution of plasmids (a) Measurement of Pteto-GFP clone on low copy number plasmid. Experimental CV=0.46, Model CV=0.32 (b) Measurement of Pteto–sfGFP-AAV cloned on low copy number plasmid. Experimental CV=0.86, Model CV=0.44. Please click here to view a larger version of this figure.
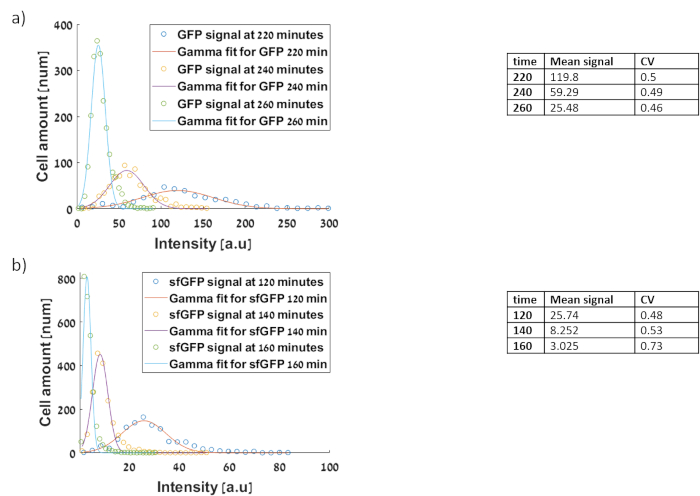
Figure 15: Histograms of the gene circuits based on microscope: experimental data (dotted lines) and stochastic model data (solid lines). The x axis represents arbitrary fluorescence units from inverted microscope and the y axis represents the frequency of cells producing the corresponding fluorescence level. Fitting was made using the MATLAB gamma distribution31,32 (a) Measurement of PtetO-GFP clone on low copy number plasmid (b) Measurement of PtetO–sfGFP-AAV cloned on low copy number. Comparison shows that our protocol obtains similar CV values as in flow analyzer experiment. Please click here to view a larger version of this figure.
Supplementary Figure 1: Four dilution ratios, between 1:500 and 1:20, for the MG1655 strain. The graph shows that if initial density is high, LB nutrients are abundant resulting in cells dividing faster. For a 1:500 dilution, cell density remained constant. For a 1:100 dilution, cell density increased slowly. For a 1:50 dilution, cell density increased at moderate division rate without significant delay in 250 RPM incubation. For a 1:20 dilution, the cell density reached saturation fast. We chose a dilution ratio of 1:30, allowing short incubation for reaching exponential growth and substantial division range for achieving high saturation at moderate division rate. Please click here to download this figure
Supplementary Figure 2: Processing bright field images. (a) Raw data intensity images from the bright field channel from the microscope system. (b) Contrast enhancement manipulation of the image to improve separation of cell objects from the background. (c) Cell boundary recognition based on convolution filter33 and binarization based global threshold34. (d) Morphological35 operations of closing and filling to identify cell colony boundaries. Please click here to download this figure
Supplementary Figure 3: Cell colony backgroundforeground segmentation. To minimize the impact of the noise we try to identify cell colony boundaries and extract them from the gel noise. (a) Image binarization operation on contrast enhancement based on adaptive thresholding to detect cell objects36. (b) Matrix multiplation of matrixes presented at S2d and S3a successfully filtering out most noises and differentiating gel noise from cells. Some boundries are not fully resolved. (c) Identification of cell boundaries using a watershed algorithm37 based on distance transform38. (d) Matrix multiplation of matrixes presented at S3b and S3c successfully resolving single cells. Please click here to download this figure
Supplementary Figure 4: Single cell segmentation. (a) Segmented product of bright field image. Further cleaning is preformed based on cell area gating and shape, discarding round bubbles. (b) Fluorescence signal images acquired from microscope. (c) Matrix multiplation of matrixes presented at S4a and S4b successfully extracting the signal of single cells. Please click here to download this figure
Supplementary Figure 5: Loss of monolayer. (a) Bright field image of a cell layer at 840 minutes (14 hours) of Pteto that regulates GFP circuit shown in figure 2 in the article as a good experiment. At the right side of microscopy image loss of monolayer can be detected. (b) Software segmentation image. Due to loss of monolayer cells are fragmented and will be cleaned base on area gating. (c) Fluorescence image showing that on the right side of image no cells can be resolved and a low signal of cells on the left side of the image due to loss of monolayer. (d) Segmentation image combined with fluorescence image. Please click here to download this figure
Supplementary Video 1. Please click here to download this video
Supplementary Video 2. Please click here to download this video
Supplementary Video 3. Please click here to download this video
cell_growth_rate_fit.m. Please click here to download this file
compare_experiments.m. Please click here to download this file
Count_Cells.m. Please click here to download this file
main_code.m. Please click here to download this file
In this work, we developed a protocol that enables computer tracing of Escherichia coli live cells, following division and fluorescent levels over a period of hours. This protocol allows us to quantify the stochastic dynamics of genetic circuits in Escherichia coli by measuring the CV and SNR in real time. In this protocol, we compared the stochastic behaviors of two different circuits as shown in Figure 10. It has been shown that plasmids with low copy numbers are more prone to stochastic effects and less affected by cell division. The first circuit constitutively expressed GFP (Figure 10a), and the second circuit constitutively expressed sfGFP fused to a ssrA degradation tag (Figure 10b). In order to quantify the stochastic behavior of the fluorescent proteins, we also recorded the bright field images. The results show that the expression of GFP, specifically at its maturation39, is the dominant noise source. The periodic saw tooth behavior pattern observed in Figure 10a can be explained by the random process of cell division and the long-time scale of GFP maturation (~50 minutes). By contrast, the sfGFP signal of the second circuit was stabilized during the measurement, because the very short maturation time of the sfGFP (~6 minutes). We developed a simple formula that describes the level of GFP in both circuits when we consider only the process of cell division and GFP maturation time. At a given time, t, we assume that there are x copy numbers of proteins, and µ copy numbers of plasmids. The protein copy number can be described by:
1.3 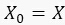
1.4 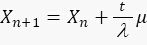
When considering only division events; 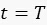 after protein maturation and so for the specific plasmids we get:
after protein maturation and so for the specific plasmids we get:
For GFP (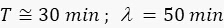 ): 1.5
): 1.5 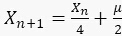
For sfGFP (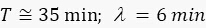 ):1.6
):1.6 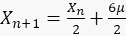
Then, the developed series of sfGFP:
1.7 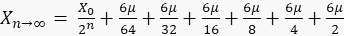
In the case that 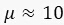 , we obtain
, we obtain 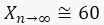 . This result can be explained as follows. When x is small, sfGFP levels increase by a small amount. When x is large, sfGFP is degraded to a steady state. Similarly, for the circuit of GFP, each cell contains about 10 units of GFP, but since maturation time for GFP is longer than the mitosis, degrading saw tooth patterns of fluorescence intensities are observed. In the model, we assumed that replication of the plasmid is fast enough that plasmid distribution remains constant before and after division. The ssrA degradation tag often reduces the protein half-life time from hours to less than an hour5 and leads to a fast steady state. We showed that the method also provides a distribution similar to that measured with a high population flow analyzer and that the CV of this distribution is equal to or smaller than the flow analyzer CV.
. This result can be explained as follows. When x is small, sfGFP levels increase by a small amount. When x is large, sfGFP is degraded to a steady state. Similarly, for the circuit of GFP, each cell contains about 10 units of GFP, but since maturation time for GFP is longer than the mitosis, degrading saw tooth patterns of fluorescence intensities are observed. In the model, we assumed that replication of the plasmid is fast enough that plasmid distribution remains constant before and after division. The ssrA degradation tag often reduces the protein half-life time from hours to less than an hour5 and leads to a fast steady state. We showed that the method also provides a distribution similar to that measured with a high population flow analyzer and that the CV of this distribution is equal to or smaller than the flow analyzer CV.
While several methods3,7,18 were developed for live cell imaging, the method presented here is tailored specifically to Escherichia coli. This bacterium requires a special media and a slightly different approach. The protocol has the following important features: (1) Establishing a dilution ratio specific to the bacteria and strain at the beginning of exponential growth rather than at the middle stage (Exponential growth without shaking) (2) Escherichia coli best divide at 37 °C, but at 37 °C the minimal media gel loses water rapidly which leads to shrinkage and instability. The protocol here overcomes this challenge (3) We first apply the bacteria sample and then seal it with liquid gel. Since the sample is trapped between the glass bottom and gel we require a gel that (I) allows gas exchange to avoid cutting air pockets, (II) remains liquid at low temperatures as Escherichia coli are sensitive to heat and (III) makes sure the sample will not be mixed inside the liquid gel. The gel remains liquid at 37 °C, however, we recommend use of a protective cap on top of the sample to avoid excessive heat and sample mixing inside the gel (4) This approach requires simple, generic equipment (5) Samples can be measured directly without preheating, so there is no loss of division events (6) The protocol, which includes the wet-lab steps and the customized automated software, can be used to study the stochastic behavior of genetic circuits such as quantifying the SNR and CV of circuit signals. We compared the microscopy method to flow analyzer results in order to validate the use of small populations of cells and establish a baseline for comparison according to CV (7) The software allows us to detect cells on the monolayer without human intervention and analyze total noise. Software tools such as ImageJ18 or Schnitzcells require manual identification of cells and are challenging for adjustments.
When continuously imaging living cell colonies, design the experiment while considering several complex physical parameters, such as cellular stress and toxicity, rate of resources depletion, necrosis and degradation time of inducers and chemicals. The protocol allows reliable measurement of up to five hours. Our experiments suggest that Escherichia coli synchronize when dividing in micro, monolayer colonies (Supplementary Video 3). We assume that the gel is uniform in terms of resources and toughness, the cells have similar behavior, and the field of illumination is slightly bigger from the field of view. Thus, each cell should produce the same signal and statistical data, which can be collected as we have shown by comparing CV obtained in this way to measurement with a flow analyzer. In order to measure noise at constant initial conditions for longer periods of time, we will consider using microfluidic chips in the future. Further benefits of such a device are maintaining fixed positions of cells and stable focus10. Still, design, fabrication of microfluidic chips and priming for experiments require training, specific equipment (custom made at times) and time. For this reason, it is beneficial to use time lapse microscopy as shown in this protocol to acquire a general understanding of the circuit.
The proposed protocol and the developed software allow reproducible measurements, from which it is simple to derive graphs. It also allows testing and comparison of total noise and can be modified for measuring intrinsic and extrinsic noise. The method is based on generic or easy to order materials, openly shared, easy to use software and does not require specific training. We have shown that the population measured by microscopy is big enough to obtain meaningful data by comparing the method CV to that obtained with a flow analyzer. Therefore, we can successfully establish a SNR baseline using this protocol and compare it to more complex gene circuits.
