Menschliche Zellen sind ständig einer Vielzahl von DNA-schädigenden Wirkstoffen unterschiedlicher Herkunft ausgesetzt. Exogene Quellen bestehen hauptsächlich aus der Exposition gegenüber Strahlungen, Chemikalien (einschließlich Chemotherapeutika und einigen Antibiotika) und Viren, während die wichtigsten endogene Quellen Fehler bei der DNA-Replikation und oxidativem Stress umfassen. Die direkten Auswirkungen einer genotoxischen Exposition können je nach Stress und Expositionsdosis von einer modifizierten Basis bis hin zu einem potenziell tödlichen DNA-Doppelstrangbruch (DSB) reichen. Letztlich können nicht reparierte oder falsch reparierte DNA-Schäden zur Anhäufung von Mutationen, genomischen Umlagerungen, Genominstabilität und schließlich zu Karzinogenese1führen. Säugetierzellen haben komplexe Wege entwickelt, um bestimmte Arten von DNA-Schäden zu erkennen2,3 und reparieren sie rechtzeitig, synchronisiert mit dem Zellzyklusprogression.
Ionisierende Strahlung (IR) schädigt die DNA-Doppelhelix und erzeugt Doppelstrangbrüche (DSBs), eine der schädlichsten Formen von DNA-Schäden. Der MRN-Komplex (MRE11, RAD50, NBS1) fungiert als Sensor der DNA endet und aktiviert die Proteinkinase ataxia telangiectasia mutiert (ATM)4,5. Nach der ersten Aktivierung von ATM durch DNA-Ende löst ATM eine Kaskade von DDR-Ereignissen am Ort der Pause aus, die mit einem Schlüsselereignis die Phosphorylierung der Histonvariante H2AX6einlöst. Die H2AX-Phosphorylierung auf Rückstand S139 aktiviert sie in die Region H2AX und spannt Regionen bis zu Megabasen um die DNA-Läsion6,7,8,9. Dieses Ereignis erhöht die Zugänglichkeit der DNA, was zur Rekrutierung und Akkumulation anderer DNA-Reparaturproteine führt7. Da h2AX reichlich und spezifisch um d.B. heruminduziert ist, kann es mit spezifischen Antikörpern leicht visualisiert werden und wird häufig als Ersatzmarker für DSBs im DNA-Reparaturfeld verwendet. Sobald der Bruch signalisiert ist, aktivieren Zellen ihre DNA-Reparaturwege und verarbeiten die DNA-Schäden. Das Protein MDC1 (Mediator des DNA-Schadens-Checkpoint-Proteins101) bindet direkt mit AtM11 und auch mit NBS112,13. Es trägt dazu bei, die Konzentration des MRN-Komplexes am DSB zu erhöhen und eine positive ATM-Rückkopplungsschleife zu starten. H2AX wird nach der Reparatur des Bruchs schnell entfernt, so dass die DSB-Freigabe überwacht werden kann. Gefolgt von der Mikroskopie, bietet die Abnahme von H2AX im Laufe der Zeit eine indirekte Messung von Restbrüchen und DNA-Reparatureffizienz.
Eukaryotische Zellen können DSBs durch mehrere Wege reparieren, wobei die beiden wichtigsten nicht-homologe Endverbindung (NHEJ) und homologe Rekombination (HR)(Abbildung 1) sind. NHEJ liiert im Wesentlichen DNA-Doppelstrangenden ohne die Verwendung von erweiterter Homologie und arbeitet im gesamten Zellzyklus14,15. HR überwiegt in S- und G2-Phasen und wird ansonsten unterdrückt, da es ein Schwesterchromatid als homologe Schablone für die Reparatur14,16benötigt. Die Wahl des Weges zwischen NHEJ und HR hängt nicht nur von der physischen Nähe des Schwesterchromatids ab, sondern auch von der Ausdehnung der DNA-Endresektion17, die NHEJ hemmt.
Die homologieabhängige DSB-Reparatur initiiert durch nukleolytische Degradation des 5′-Strangs von den Bruchenden, um 3′ einsträngige DNA-Schwänze (ssDNA) zu erzeugen, ein Prozess, der als 5′-3′-Resektion bezeichnet wird. Der MRN-Komplex initiiert die DNA-Endresektion und weitere Resektion wird in Kombination mit BLM/EXO1 (Bloom-Syndrom-Protein/Exonuklease 1) oder BLM/DNA2 (DNA-Replikation ATP-abhängige Helicase/Nuklease)18,19,20,21,22verarbeitet. Die DNA-Endresektion wird durch CtIP (CtBP-interagierendes Protein) durch seine direkte Interaktion mit dem MRN-Komplex23 und die Rekrutierung von BRCA1 (Brustkrebs Typ 1 Anfälligkeitsprotein)24,25verstärkt. Replikationsprotein A (RPA) bindet sich prompt an die ssDNA exponiert und wird dann durch das rekombinante Protein RAD51 zu einem Nukleoprotein-Filament verdrängt, das homologe Suche und Stranginvasion katalysiert26,27,28.
Die Einleitung der Resektion ist ein entscheidender Schritt für die Reparaturwegwahl. Sobald die Resektion eingeleitet wurde, werden die DNA-Enden zu schlechten Substraten für die Bindung durch Ku70/Ku80-Heterodimer (Komponente des NHEJ-Signalwegs) und Zellen werden HR17,29,30verschrieben. Der Ku70/Ku80 Heterodimer bindet an DSB-Enden und rekrutiert DNA-PKcs und p53 Binding Protein 1 (53BP1)29,30. 53BP1 wirkt als Hemmungshindernis in G1 und blockiert damit HR beigleichzeitigerFörderung von NHEJ 31,32, wird aber in der S-Phase in BRCA1-abhängiger Weise entfernt, so dass eine Resektion33,34möglich ist. Daher spielen 53BP1 und BRCA1 bei der DSB-Reparatur eine gegensätzliche Rolle, wobei 53BP1 ein NHEJ-Moderator ist, während BRCA1-Akte Pausen für die Reparatur durch HR ermöglichen.
Im Labor kann die DSB-Bildung durch ionisierende Strahlung (IR) induziert werden. Während dieses Beispiel eine hohe Dosis von 4 Gy verwendet, 1 Gy und 2 Gy auch eine signifikante Menge an DSBs erstellen, geeignet für die Analyse der Foci-Bildung durch reichliche Proteine. Es ist wichtig zu beachten, dass die Art und Dosis der verwendeten Strahlung zu verschiedenen Läsionen in der DNA und in der Zelle führen kann: Während IR DSBs induziert, kann es auch einzelne Strangbrüche oder Base-Modifikation verursachen (siehe35,36 für einen Verweis auf bestrahlung lineare Energieübertragung (LET) und Art der DNA-Schäden). Um die Kinetik der ionisierenden strahlungsinduzierten Brennweite (IRIF) Bildung und deren Clearance zu bestimmen, die auf die Reparatur des Schadens und die Umkehrung der aktivierten DDR8,9,37,38hinweist, kann die Brennpunktbildung zu verschiedenen Zeitpunkten nach ionisierender Strahlung überwacht werden. Timing der Aktivierung und Clearance aller wichtigen DNA-Schäden Proteine ist bekannt39, und viele werden als Ersatzmarker für Schlüsselereignisse untersucht. Beispielsweise wird pRPA, das eine hohe Affinität zu ssDNA besitzt, als Ersatz für die Bruchresektion verwendet, MRN-Proteine (MRE11, RAD50, NBS1) und Exonukleasen können auch verwendet werden, um die Resektionseffizienz zu bewerten. Während RAD51, BRCA1, BRCA2 (Brustkrebs Typ 2 Anfälligkeitsprotein) und PALB2 (Partner und Lokalisierer von BRCA2) überwacht werden können, um die HR-Effizienz zu bewerten, wird das Vorhandensein der Ku-Proteine oder 53BP1 als Marker von NHEJ(Abbildung 1)verwendet.
Während sich Proteine der DNA-Reparaturmaschinen gegenseitig bis zum Bruch rekrutieren und in Superkomplexen zusammensetzen, können DNA-Protein- und Protein-Protein-Wechselwirkungen abgeleitet werden, indem man ihrer individuellen Lokalisierung im Laufe der Zeit folgt und die Kolokalisierung von Proteinen analysiert, wie durch überlappende Signale in Zelle40,41,42visualisiert. In Zelllinien ermöglicht die Einführung von Punktmutationen oder das Löschen in bestimmten DNA-Reparaturgenen entweder durch Genombearbeitung oder durch Überexpression plasmidbasierter Mutanten die Untersuchung spezifischer Rückstände und ihre mögliche Rolle bei der Erkennung von DNA-Schäden (z. B. Kolokalisierung mit H2AX) oder komplexer Montage (Kolokalisierung mit einem anderen oder mehreren Proteinen) sowie deren Auswirkungen auf die DNA-Reparatur. Hier verwenden wir die indirekte Immunfluoreszenz als Mittel, um die Bildung und Auflösung von DSBs zu untersuchen, indem wir im Laufe der Zeit den H2AX-Brennpunkten folgen. Wir stellen auch ein Beispiel für die Foci-Bildung und Co-Lokalisierungsanalyse durch einen wichtigen Akteur in der DSB-Reparatur vor: p53 Binding Protein 1 (53BP1)32. Wie bereits erwähnt, 53BP1 gilt als zentral für DNA-Reparatur-Pfad Wahl. Nach 53BP1 Akkumulation und seine Co-Lokalisierung mit H2AX liefert wertvolle Informationen über Zellzyklusphase, DNA-Schadensakkumulation, und Weg verwendet, um DSBs zu reparieren. Der Zweck der indirekten Immunlokalisierung besteht darin, die Effizienz der REPARATUR von DNA-Schäden in Zelllinien nach IR wie in dieser Studie oder nach Exposition gegenüber verschiedenen Belastungen in der Zelle von der DNA-Vernetzung bis zur Blockierung der Replikationsgabel zu bewerten (eine Liste der DNA-schädlichen Mittel ist in Tabelle 1enthalten).
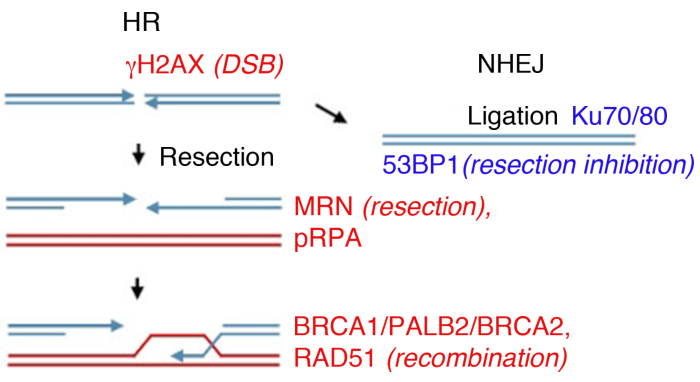
Abbildung 1: DNA Double Strand Breaks (DSB) Reparaturwege.
Die DSB-Reparatur umfasst zwei Hauptwege: Homologe Rekombination (HR, links) und Non-Homologous End-Joining (NHEJ, rechts). Nach der Pause werden Proteine aktiviert, um den Bruch zu markieren (H2AX), an der Endresektion (MRN) teilnehmen, die resezierte ssDNA (pRPA) beschichten, die Rekombination fördern (BRCA1, PALB2, BRCA2, RAD51) oder die Resektion begrenzen und NHEJ (53BP1) fördern. Andere Proteine nehmen an der Schadensreparatur teil, aber die aufgeführten Proteine werden routinemäßig mit indirekter Immunfluoreszenz gefolgt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
| DNA-schädigendes Mittel | Wirkmechanismus | Empfohlene Dosis |
| A-Strahlen/Röntgenstrahlen | Strahlung Bildung von doppelsträngigen Brüchen mit einigen unkontrollierten zellulären Effekten |
1-4 Gy |
| 36 Ar ionen | Strahlung Bildung von Doppelstrandedpausen |
270 keV/m |
| •Partikel | Strahlung Bildung von Doppelstrandedpausen |
116 keV/m |
| Bleomycin | Inhibitor der DNA-Synthese | 0,4-2 g/ml |
| Camptothecin | Inhibitor der Topoisomerase I | 10-200 nM |
| Cisplatin | Alkylierungsmittel (induzierenintrastrande Querverbindungen) |
0,25-2 m |
| Doxorubicin | Intercalating-Agent Inhibitor der Topoisomerase II |
10-200 nM |
| Etoposide | Inhibitor der Topoisomerase II | 10 m |
| Hydroxyurea | Inhibitor der DNA-Synthese (durch Ribonukleotid-Reduktase) |
10-200 m |
| Methylmethansulfonat | Alkylierungsmittel | 0,25-2 mM |
| Mitomycin C | Alkylierungsmittel | 0,25-2 m |
| Ultraviolettes (UV) Licht | Bildung von Thymidin-Dimeren (Erzeugung von Verzerrungen der DNA-Kette) |
50-100 mJ/cm2 |
Tabelle 1: Genotoxische Mittel. Beispiele für DNA-schädigende Mittel, deren Wirkmechanismus und der verursachte Schaden auf der Grundlage der vorgeschlagenen Arbeitskonzentration.
