Las células humanas están constantemente expuestas a una variedad de agentes dañinos del ADN de diversos orígenes. Las fuentes exógenas consisten principalmente en la exposición a radiaciones, productos químicos (incluidos agentes quimioterápticos y algunos antibióticos) y virus, mientras que las principales fuentes endógenas incluyen errores en la replicación del ADN y el estrés oxidativo. Los efectos directos de la exposición genotóxica pueden variar de una base modificada a una rotura de doble cadena de ADN potencialmente letal (DSB), dependiendo de la tensión y la dosis de exposición. En última instancia, el daño del ADN no reparado o mal reparado puede conducir a la acumulación de mutaciones, reordenamientos genómicos, inestabilidad del genoma y eventualmente conducir a la carcinogénesis1. Las células de mamíferos han evolucionado vías complejas para reconocer tipos específicos de daño del ADN2,3 y repararlos de manera oportuna, sincronizados con la progresión del ciclo celular.
La radiación ionizante (IR) daña la doble hélice del ADN y crea roturas de doble cadena (DSB), una de las formas más perjudiciales de daño al ADN. El complejo MRN (MRE11, RAD50, NBS1) funciona como un sensor de ADN termina y activa la proteína quinasa ataxia telangiectasia mutada (ATM)4,5. Después de la activación inicial de ATM por los extremos de ADN, ATM desencadena una cascada de eventos DDR en el sitio de la rotura, iniciando con un evento clave, la fosforilación de la variante de histona H2AX6. La fosforilación H2AX en el residuo S139 la activa en el OH2AX, abarcando regiones hasta megabases alrededor de la lesión de ADN6,7,8,9. Este evento aumenta la accesibilidad del ADN, lo que lleva al reclutamiento y acumulación de otras proteínas de reparación del ADN7. Debido a que el S2AX es abundante y específicamente inducido por DSB, se puede visualizar fácilmente utilizando anticuerpos específicos, y se utiliza comúnmente como un marcador suplente para DSB en el campo de reparación del ADN. Una vez que se señala la rotura, las células activan sus vías de reparación del ADN y procesan el daño del ADN. La proteína MDC1 (mediador de la proteína de control de daño del ADN 1) se une directamente a la proteína de control de daño del ADN10, interactúa con el ATM11 y también con NBS112,,13. Contribuye a aumentar la concentración del complejo MRN en el OSD e iniciar un bucle de retroalimentación ATM positivo. En consecuencia, se elimina rápidamente el H2AX una vez que se repara la rotura, lo que permite la supervisión del aclaramiento del OSD. Seguido de la microscopía, la disminución de la V2AX con el tiempo proporciona una medición indirecta de las roturas residuales y la eficiencia de la reparación del ADN.
Las células eucariotas pueden reparar DSB por varias vías, siendo las dos principales la unión final no homóloga (NHEJ) y la recombinación homóloga (HR) (Figura 1). NHEJ esencialmente liga los extremos de doble hebra de ADN sin el uso de homología extendida y opera a lo largo del ciclo celular14,15. HR se vuelve predominante durante las fases S y G2, y de lo contrario se reprime, ya que requiere un cromátido hermano como plantilla homóloga para la reparación14,16. La elección de la vía entre NHEJ y HR no sólo depende de la proximidad física del cromátido hermano, sino también de la extensión de la resección final del ADN17,que inhibe el NHEJ.
La reparación del DSB dependiente de la homología inicia por degradación nucleolítica de la hebra de 5′ desde los extremos de rotura para generar colas de ADN de una sola cadena (ssDNA) de 3′, un proceso denominado resección de 5′-3′. El complejo MRN inicia la resección del extremo del ADN y la resección posterior se procesa en combinación con BLM/EXO1 (proteína del síndrome de Bloom/exonucleasa 1) o BLM/DNA2 (helicasa/nucleasa dependiente de la replicación del ADN)18,19,20,21,22. La resección final del ADN se ve reforzada por CtIP (proteína que interactúa con CtBP) a través de su interacción directa con el complejo MRN23 y el reclutamiento de BRCA1 (proteína de susceptibilidad tipo 1 de cáncer de mama)24,,25. La proteína de replicación A (RPA) se une rápidamente al ssDNA expuesto y luego es desplazada por la proteína recombinase RAD51 para formar un filamento de nucleoproteína que cataliza la búsqueda homóloga y la invasión dehebras 26,,27,,28.
El inicio de la resección es un paso crítico para la elección de la vía de reparación. Una vez iniciada la resección, los extremos del ADN se convierten en sustratos pobres para la unión por heterodista Ku70/Ku80 (componente de la vía NHEJ) y las células se comprometen a HR17,,29,,30. El heterodimero Ku70/Ku80 se une a los extremos del OSD, reclutando DNA-PKcs y p53 Binding Protein 1 (53BP1)29,30. 53BP1 actúa como barrera de resección en G1, bloqueando así los recursos humanos mientras se promueve NHEJ31,32, pero se elimina de forma dependiente de BRCA1 en la fase S, permitiendo así que se produzca la resección33,,34. Por lo tanto, 53BP1 y BRCA1 desempeñan funciones opuestas en la reparación del OSD, siendo 53BP1 un facilitador DE NHEJ, mientras que BRCA1 actúa permitiendo la reparación a través de HR.
En el laboratorio, la formación de DSB puede ser inducida por radiación ionizante (IR). Mientras que este ejemplo utiliza una dosis alta de 4 Gy, 1 Gy y 2 Gy también crean una cantidad significativa de DSBs, adecuado para el análisis de la formación de focos por proteínas abundantes. Es importante tener en cuenta que el tipo y la dosis de radiación utilizada pueden conducir a diferentes lesiones en el ADN y en la célula: mientras que ir ir induce DSB, también puede causar roturas de una sola hebra o modificación de la base (ver35,36 para una referencia sobre la irradiación de transferencia de energía lineal (LET) y el tipo de daño del ADN). Para determinar la cinética de la formación de focos inducidos por radiación ionizante (IRIF) y su aclaramiento, que indican la reparación del daño y la reversión de la ACTIVA DDR8,9,37,38, la formación de focos puede ser monitoreada en diferentes puntos de tiempo después de la radiación ionizante.38 El tiempo de activación y aclaramiento de todas las proteínas de daño del ADN importante se conoce39, y muchos se investigan como marcadores sustitutos de eventos clave. Por ejemplo, pRPA, que posee una alta afinidad por ssDNA se utiliza como sustituto de la resección de rotura, las proteínas MRN (MRE11, RAD50, NBS1) y las exonucleas también se pueden utilizar para evaluar la eficiencia de la resección. Mientras que RAD51, BRCA1, BRCA2 (proteína de susceptibilidad tipo 2 de cáncer de mama), y PALB2 (socio y localizador de BRCA2) se pueden monitorear para evaluar la eficiencia de HR, la presencia de las proteínas Ku o 53BP1, se utilizan como marcadores de NHEJ (Figura 1).
A medida que las proteínas de la maquinaria de reparación del ADN se reclutan entre sí para la ruptura y se ensamblan en supera complexios, las interacciones ADN-proteína y proteína-proteína se pueden inferir siguiendo su localización individual a lo largo del tiempo y analizando la co-localización de proteínas, visualizadas por señales superpuestas en las células40,,41,,42. En las líneas celulares, la introducción de mutaciones puntuales o la deleción en genes específicos de reparación del ADN, ya sea a través de la edición del genoma, o mediante la sobreexpresión de mutantes a base de plásmidos, permite la investigación de residuos específicos y su posible papel en el reconocimiento del daño del ADN (por ejemplo, la co-localización con el OH2AX) o el montaje complejo (co-localización con otra, o varias, proteínas), así como su impacto en la reparación del ADN. Aquí, utilizamos la inmunofluorescencia indirecta como medio para investigar la formación y la resolución de los DSB siguiendo a los focos de H2AX a lo largo del tiempo. También presentamos un ejemplo de formación de focos y análisis de co-localización por un actor importante en la reparación del DSB: p53 Binding Protein 1 (53BP1)32. Como se mencionó anteriormente, 53BP1 se considera fundamental para la elección de la vía de reparación del ADN. Después de la acumulación de 53BP1 y su co-localización con el S2AX proporciona información valiosa sobre la fase del ciclo celular, la acumulación de daño en el ADN y la vía utilizada para reparar DSB. El propósito de la inmunolocalización indirecta es evaluar la eficiencia de la reparación del daño del ADN en las líneas celulares, después de IR como en este estudio, o después de la exposición a diversas tensiones en la célula, desde la reticulación del ADN hasta el bloqueo de la horquilla de replicación (se proporciona una lista de agentes que dañan el ADN en la Tabla 1).
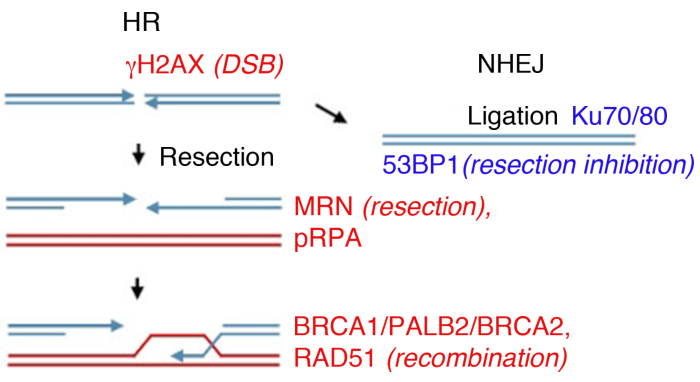
Figura 1: Dna double strand breaks (DSB) vías de reparación.
La reparación del DSB implica dos vías principales: recombinación homóloga (HR, izquierda) y unión final no homóloga (NHEJ, derecha). Después de la rotura, las proteínas se activan para marcar la rotura ( H2AX), participar en la resección final (MRN), recubrir el ssDNA resecado (pRPA), promover la recombinación (BRCA1, PALB2, BRCA2, RAD51) o limitar la resección y promover NHEJ (53BPBP). Otras proteínas participan en la reparación del daño, pero las proteínas enumeradas son seguidas rutinariamente por inmunofluorescencia indirecta. Haga clic aquí para ver una versión más grande de esta figura.
| Agente dañante del ADN | Mecanismo de acción | Dosis recomendada |
| Rayos/rayos X | Radiación Formación de roturas de doble cadena con algunos efectos celulares incontrolados |
1-4 Gy |
| 36 Incendios | Radiación Formación de roturas de doble cadena |
270 keV/m |
| partículas | Radiación Formación de roturas de doble cadena |
116 keV/m |
| Bleomicina | Inhibidor de la síntesis de ADN | 0,4-2 g/ml |
| Camptotecina | Inhibidor de la topoisomerasa I | 10-200 nM |
| Cisplatino | Agente alquilante (induciendo reticulaciones intrastrand) |
0,25-2 M |
| Doxorrubicina | Agente intercalador Inhibidor de la topoisomerasa II |
10-200 nM |
| Etoposide | Inhibidor de la topoisomerasa II | 10 M |
| Hidroxiurea | Inhibidor de la síntesis de ADN (por ribonucleótido reductasa) |
10-200 m |
| Metanoesulfonato de metilo | Agente alquilante | 0,25-2 mM |
| Mitomicina C | Agente alquilante | 0,25-2 M |
| Luz ultravioleta (UV) | Formación de dimers de timidina (generando distorsión de la cadena de ADN) |
50-100 mJ/cm2 |
Tabla 1: Agentes genotóxicos. Ejemplos de agentes dañinos para el ADN, su mecanismo de acción y el daño inducido en función de la concentración de trabajo sugerida.
