Immunostaining and imaging of the whole ovary enables the visualization and quantification of oocytes or protein expression in ovaries at different developmental stages using the same technique and markers (Figure 3). This protocol was developed for a large-scale project in which analysis of ovaries at multiple stages and from multiple mouse strains was required. Here, we present data gathered for the C57BL6/J strain, a standard strain for genetic analysis. The technique presented here is straightforward, results can be obtained within 14-19 days (Figure 2D) and can be used for ovaries from prenatal, prepubertal, and pubertal females (Figure 1 and Figure 2A,B).
This approach was used to study the dynamics of the oocyte loss that occurs naturally during the formation of the ovarian oocyte reserve. In many organisms including the mouse, oocyte numbers are thought to peak during fetal life around the time that oocytes enter meiosis ~E13.5. Oocyte numbers decrease due to the still-not-fully-understood process of fetal oocyte attrition (FOA) (from ~E15.5 to E18.5), which has been mechanistically linked to the expression of the retrotransposon LINE-120,21. More oocytes are eliminated after E18.5 to P0 due to the elimination of abnormal oocytes by a meiotic quality checkpoint22,23. To investigate these processes using this protocol, we immunostained and imaged ovaries at different developmental stages using DDX4 and GCNA as oocyte markers as shown in Figure 3.
Small oocytes expressing GCNA and DDX4 are seen from E15.5 onwards and they represent oocytes during MPI or arrested at dictyate within primordial follicles. Larger growing oocytes with strong DDX4 expression are detected already in P2 ovaries where they most likely represent the first wave of follicles44. Increasing numbers of larger oocytes are seen in P5 and P28 ovaries. Images obtained by multiphoton microscopy (Figure 4A) were processed using IMARIS software and 3D rendering was performed to identify small and larger growing oocytes based on the nuclear GCNA signal and the size delineated by the DDX4 signal (Figure 4A and Video 1). Oocytes were counted as described in the protocol and results are summarized in Figure 4B. In agreement with previous studies, we observed a significant oocyte loss from E15.5 to E18.5 (~32%) and E18.5 to P2 (~24%). By the time females reach puberty (P28), only ~30% of oocytes present at E15.5 have survived.
This method can be also used to observe the consequences of genotoxic treatments such as irradiation, which has been shown to completely eliminate the primordial follicle reserve within one week23,38,45. A significant visual difference is evident between the whole ovary treated with radiation and the untreated control in Figure 3 (comparing P28 ovaries from treated and untreated females). In the P28 ovary without radiation exposure, we observed two oocyte populations labeled with DDX4; abundant small oocytes in primordial follicles (closed arrow); and larger oocytes of various sizes typically found in growing follicles (e.g., primary, secondary, and preantral) (Figure 3). In contrast, the ovary from a female exposed to 0.5Gy of γ-radiation at P7 is completely devoid of small oocytes in primordial follicles as previously observed in 2D sections. Interestingly, only larger oocytes of similar size survived radiation; these may be the first wave follicles already growing in the P7 ovary, as they are known to be resistant to radiation46 (Figure 3, P28).
In addition to the quantification of oocyte numbers, this protocol can be used for immunostaining and quantitative analyses of other proteins involved in oocyte development. For example, we used antibodies against LINE-1ORF1p protein, an RNA-binding chaperone protein, produced by LINE-1 retrotransposons (Figure 5). Increase in the abundance of LINE-1 elements from E15.5 to E18.5 has been proposed to cause oocyte elimination20,21. Indeed, using multiphoton-captured images and signal intensity analysis in IMARIS, LINE-1 ORF1p levels were observed to increase significantly in oocytes during this time, which correlates with a significant drop in oocyte numbers as shown in Figure 4B.
In circumstances where a part of an ovary is damaged or lost, ovaries from the same strain and developmental stage can be used to computationally estimate the number of oocytes within the missing region as shown in Figure 6. Data from E15.5 ovaries were used for simulations to test the accuracy of computational corrections. Oocytes in ovaries with up to 30% tissue damage can be computationally estimated to have ≤10% deviation compared to oocytes in an intact ovary, suggesting that 3D ovary staining and modeling can be effectively used to salvage data from precious tissues. Simulations performed with ovaries at E15.5 indicate that correcting for a ≥30% loss results in a significant deviation from actual numbers (Figure 6C).
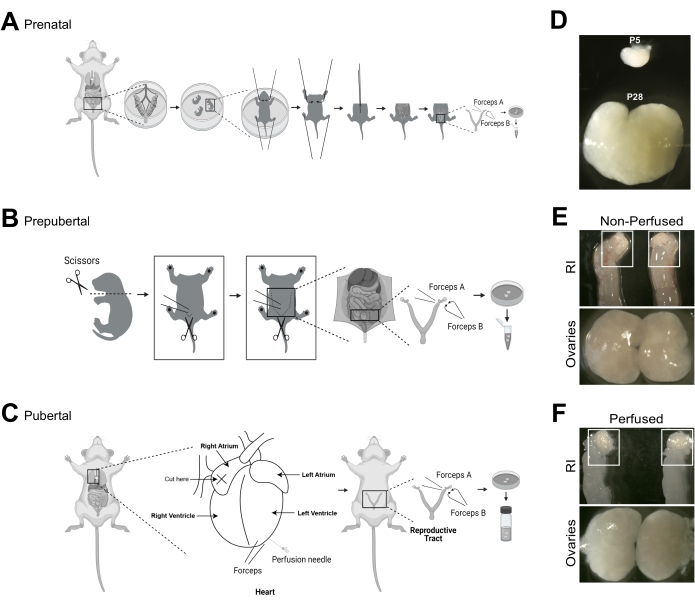
Figure 1: Ovary dissection and perfusion from females from prenatal and postnatal stages. (A–C) Ovary dissection from different stages requires different techniques, which are depicted schematically to complement the descriptions in the video. (D) Ovaries at postnatal day 5 (P5) are much smaller than at P28, which necessitates a different clearing protocol as described in the protocol. (E,F) Proper perfusion of the ovaries is important to eliminate background staining from red blood cells. Non-perfused reproductive tracts and ovaries have pink hue while perfused organs will turn white. Abbreviation: RI = non-perfused reproductive tracts. Images A–C were created with BioRender.com. Please click here to view a larger version of this figure.
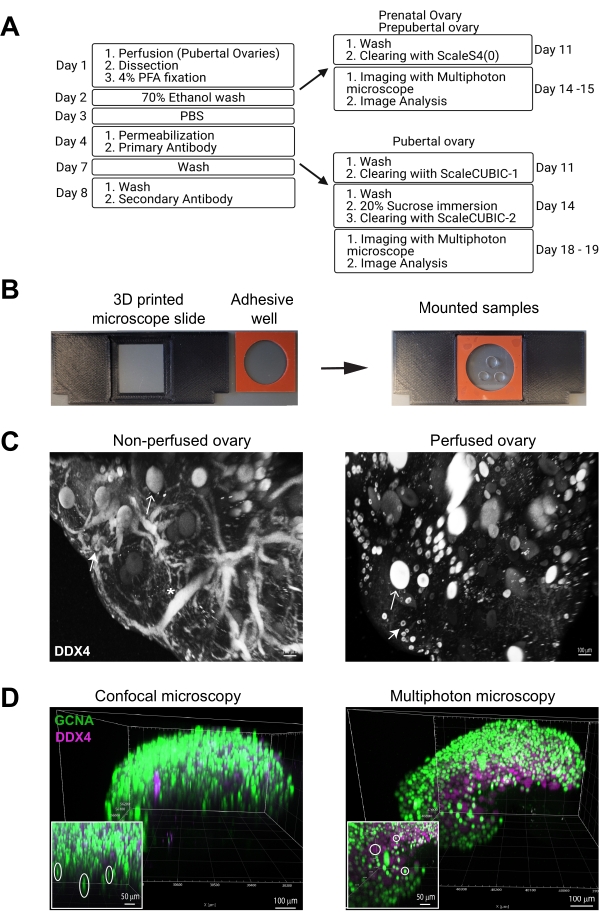
Figure 2: Immunostaining, clearing, and imaging of whole mouse ovaries. (A) Flow chart depicts shared and specific steps for immunostaining and clearing protocol for prenatal/prepubertal and pubertal ovaries. (B) Cleared ovaries are mounted in a drop of clearing solution in the middle of an adhesive well sandwiched between two cover slips. For imaging, the mounted samples are placed in a 3D printed adaptor slide. (C) Greyscale multiphoton images of non-perfused vs. perfused ovaries, immunostained with the oocyte marker DDX4, showing improved image quality and lower nonspecific staining after perfusion. The closed arrow indicates an oocyte with a thin layer of cytoplasmic DDX4 staining typical for primordial follicles, and the open arrows show larger oocytes within growing follicles. Asterisk indicates autofluorescence from blood vessels. (D) 3D renders from confocal vs. multiphoton images of P5 ovaries, immunostained with oocyte markers GCNA (green) and DDX4 (magenta), showing a significant spherical aberration from confocal microscopy and virtually none from multiphoton. Scale bars = 100 µm (C, D) and 50 µm (insets in D). Abbreviations: GCNA = germ cell nuclear acidic peptidase; DDX4 = DEAD-box helicase 4. Image A was created with BioRender.com. Please click here to view a larger version of this figure.
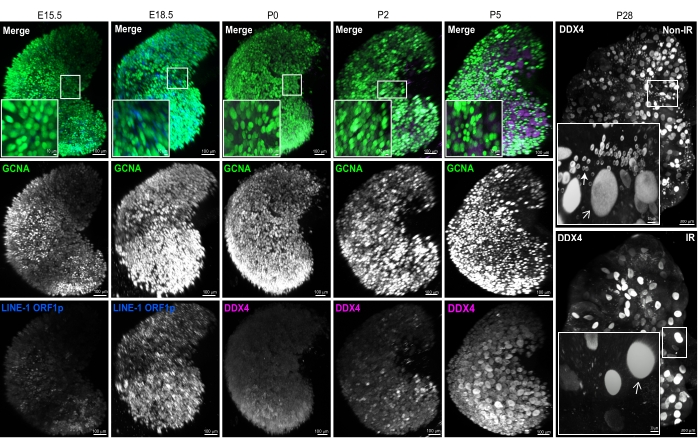
Figure 3: Representative 3D-rendered images of ovaries of different developmental stages. 3D renderings from multiphoton images of whole mount ovaries immunostained with GCNA (green) and LINE-1 ORF1p (blue) or DDX4 (magenta), in prenatal and postnatal ovaries, respectively. Individual channels are presented in grayscale to show nuclear and cytoplasmic signals. The white boxes outline regions magnified in the left bottom insets. Two different ovaries are shown for P28. The ovary from the control non-irradiated female (top) contains a large population of small oocytes in primordial follicles (see inset). In contrast, the ovary from the female irradiated at P7 with 0.5 Gy of γ-radiation (IR) is completely devoid of small oocytes in primordial follicles (see inset). The closed arrow indicates an oocyte with a thin layer of cytoplasmic DDX4 staining typical for primordial follicles, and open arrows show larger oocytes within growing follicles. Scale bars = 100 µm; 30 µm for P28 insets; 10 µm for all other insets. Insets contain magnified views of 3D-rendered images generated in IMARIS and may differ slightly from low-magnification images due to perspective. Abbreviations: Non-IR = control non-irradiated; IR = irradiated at P7 with 0.5 Gy of γ-radiation; GCNA = germ cell nuclear acidic peptidase; DDX4 = DEAD-box helicase 4; LINE-1 = long interspersed nuclear element-1. Please click here to view a larger version of this figure.
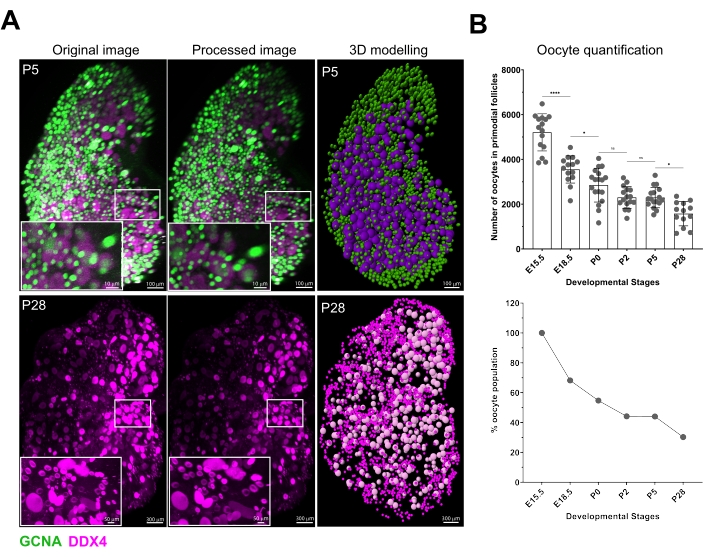
Figure 4: Image processing, 3D display of oocytes, and quantification results. (A) 3D renders from multiphoton images (left) were processed in IMARIS using Gaussian filter (middle) and oocytes of small and large sizes were identified using the spot feature (right). P5 and P28 ovaries shown as example. Scale bars = 100 µm (P5); 10 µm (P5 insets); 300 µm (P28); 50 µm (P28 insets). (B) Small oocytes positive for GCNA were quantified in ovaries from different stages using spot features (top). To illustrate decreasing numbers of oocytes during development, the average % of oocytes were calculated at each stage as compared to the average number present in the earliest stage counted at E15.5 (bottom). Note the large drop in oocyte numbers from E15.5 to E18.5. Data are presented as means ± SD. Statistical analyses were performed using GraphPad Prism software and analyzed by one-way ANOVA, and the significance was determined by Bonferroni's post hoc multiple comparison test. * P ≤ 0.05; **** P ≤ 0.0001; ns >0.05. Abbreviation: GCNA = germ cell nuclear acidic peptidase; ns = not significant. Please click here to view a larger version of this figure.
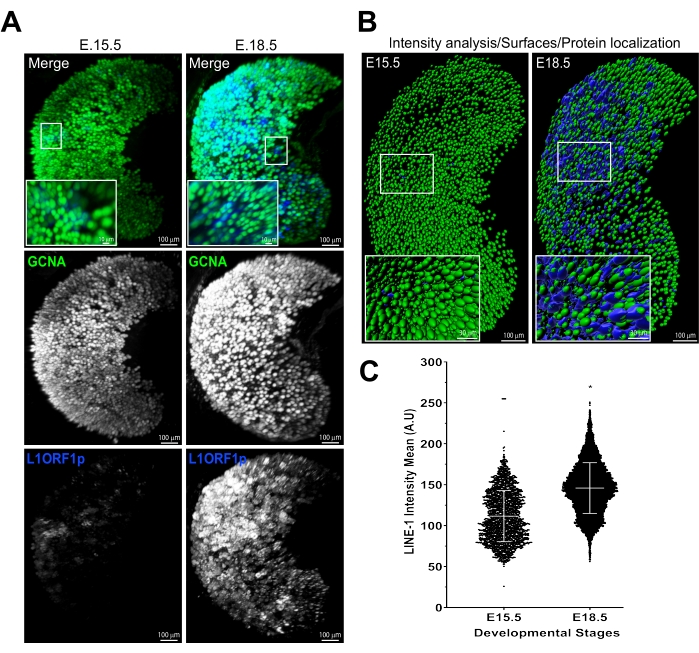
Figure 5: Detection and quantification of theLINE-1 ORF1p expression in fetal oocytes. (A) 3D renderings from multiphoton images show LINE-1 ORF1p (blue) expression in E15.5 and E18.5 fetal oocytes (marked by green GCNA). (B) 3D surfaces generated in IMARIS using top row images in panel A. (C) LINE-1 ORF1p intensity analysis shows higher expression of LINE-1 ORF1p per oocyte at E18.5 than E15.5. Scale bars = 100 µm; 10 µm (panel A insets); 30 µm (panel B insets). Data are presented as means ± SD. Statistical analyses were performed using GraphPad Prism software and analyzed by Student's t-test, and the significance was determined by Mann-Whitney U test. P ≤ 0.0001; ns >0.05. Abbreviations: GCNA = germ cell nuclear acidic peptidase; LINE-1 = long interspersed nuclear element-1. Please click here to view a larger version of this figure.
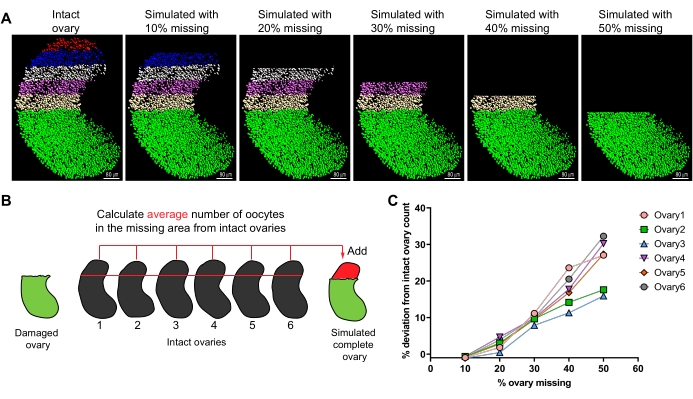
Figure 6: Method for estimating total oocyte numbers in damaged ovarian samples with computational correction. (A) Model of GCNA-positive (green) E15.5 oocytes with five 10% regions highlighted in red, blue, gray, magenta, and brown in an intact ovary. Each succeeding image, after the intact ovary, represents a simulated ovary with 10% incremental regions missing up to 50%. (B) Schematic of computational method to estimate oocyte number in damaged sample. (C) Total oocyte numbers in simulated ovaries were compared to numbers in the original intact ovaries (considered 100%) and the difference is presented as % deviation. Simulation from six individual ovaries with 10-50% volume missing. Scale bars = 80 µm. Abbreviation: GCNA = germ cell nuclear acidic peptidase. Please click here to view a larger version of this figure.
Video 1: 3D rendering and 3D modeling of oocytes in P5 ovaries. Please click here to download this Video.
| Solutions and buffers | Reagent(s) | Composition |
| Fixative | Paraformaldehyde | 4% (v/v) |
| Permeabilization buffer | Polyvinyl alcohol (PVA) | 0.2% (w/v) |
| Sodium borohydride | 0.1% (w/v) | |
| Triton X-100 | 1.5% (v/v) | |
| Blocking buffer | Bovine Serum Albumin (BSA) | 3% (w/v) |
| 1 M Glycine (pH 7.4) | 2% (w/v) | |
| Triton X-100 | 0.1% (v/v) | |
| 200x Penicillin-Streptomycin | 1% (v/v) | |
| 10% Sodium azide | 0.2% (v/v) | |
| goat serum | 10% (v/v) | |
| Washing buffer | PVA | 0.2% (w/v) |
| Triton X-100 | 0.15% (v/v) | |
| 10% Sodium azide | 0.1% (v/v) | |
| ScaleS4(0) solution (pH 8.1) | D-Sorbitol | 40% (w/v) |
| Urea | 24% (w/v) | |
| Glycerol | 10% (v/v) | |
| DMSO | 20% (v/v) | |
| ScaleCUBIC-1 solution | Urea | 25% (w/v) |
| N,N,N′,N′-Tetrakis(2-Hydroxypropyl)ethylenediamine | 25% (v/v) | |
| Triton X-100 | 15% (v/v) | |
| Sucrose Solution | Sucrose | 20%(w/v) |
| ScaleCUBIC-2 solution | Sucrose | 50% (w/v) |
| Urea | 25% (w/v) | |
| Triethanolamine | 10% (w/v) | |
| Triton X-100 | 0.1% (v/v) |
Table 1: Solutions and buffers.
Supplemental Figure S1: Image acquisition preferences. Please click here to download this File.
Supplemental Table S1: Image acquisition settings. Please click here to download this Table.
