Évaluation in vitro et in vivo de composés biologiquement actifs photocontrôlés - candidats médicaments potentiels pour la photopharmacologie du cancer
Summary
Ce protocole présente un ensemble d’expériences adoptées pour l’évaluation de peptides anticancéreux photocommutables, qui peuvent être utilisés dans le criblage préclinique de tels composés. Cela comprend l’évaluation de la cytotoxicité dans des cultures cellulaires 2D et 3D, l’évaluation de l’efficacité de la photoisomérisation ex vivo (tissu modèle) et l’efficacité in vivo .
Abstract
Les composés photocontrôlés biologiquement actifs sont une classe émergente de candidats médicaments « intelligents ». Ils offrent une sécurité supplémentaire dans la chimiothérapie systémique en raison de leur activation spatio-temporelle précise en dirigeant une lumière bénigne et non ionisable vers un endroit spécifique du corps du patient. Cet article présente un ensemble de méthodes pour évaluer la puissance in vitro et l’efficacité ex vivo de la photoactivation de composés biologiquement actifs photocontrôlés ainsi que l’efficacité in vivo aux premiers stades du développement de médicaments. La méthodologie est appliquée aux peptides cytotoxiques anticancéreux, à savoir les analogues contenant du diaryléthane d’un antibiotique connu, la gramicidine S. Les expériences sont réalisées en utilisant des cultures cellulaires 2D (cellules adhérentes) et 3D (sphéroïdes) d’une lignée cellulaire cancéreuse (carcinome pulmonaire de Lewis, LLC), de substituts de tissus vivants (hachis de viande de porc) et d’un modèle de cancer allogreffé (LLC sous-cutané) chez des souris immunocompétentes. La sélection des composés les plus efficaces et l’estimation de fenêtres photothérapeutiques réalistes sont effectuées par microscopie à fluorescence automatisée. L’efficacité de la photoactivation à différents régimes d’éclairage est déterminée à différentes profondeurs dans un tissu modèle, et le dosage optimal de la lumière est appliqué dans l’expérience thérapeutique finale in vivo .
Introduction
Les composés biologiquement actifs photocontrôlés sont apparus au cours des dernières décennies comme un composant prometteur des chimiothérapies sûres pour les maladies humaines et pour éradiquer spécifiquement les tumeurs solides malignes1. Ces composés contiennent des fragments photoisomérisables réversibles (photocommutateurs moléculaires) et peuvent basculer entre les photoisomères inactifs et actifs lors de l’irradiation avec une lumière de différentes longueurs d’onde.
Par rapport à leurs analogues non photocontrôlables, les médicaments photocontrôlés peuvent être plus sûrs car ils peuvent être introduits par voie systémique dans le corps du patient sous des formes moins actives et essentiellement non toxiques, et ne sont ensuite activés par la lumière que lorsque cela est nécessaire, comme dans les tumeurs, les ulcères et les plaies. Bien que de multiples démonstrations passionnantes de tels prototypes de médicaments moléculaires puissent être trouvées dans des articles universitaires récents 2,3,4,5,6,7, le domaine de la photopharmacologie clinique – une application de combinaisons médicament/dispositif médical/maladie approuvées – n’existe pas. La photopharmacologie est encore au stade de la découverte de médicaments et les études précliniques systématiques sont inconnues.
Ce n’est que très récemment que nous avons démontré l’avantage de sécurité in vivo pour certains peptides anticancéreux photocontrôlés, à savoir les analogues du peptide antibiotique gramicidine S8. Ces dérivés photocontrôlés contiennent un photocommutateur diaryléthène (DAE), qui subit des transformations photoinduites réversibles entre les photoformes dites « ring-open » générées par la lumière rouge et les photoformes « ring-closed » générées par UV (illustrées à la figure 1 pour l’un des dérivés, le composé LMB002).
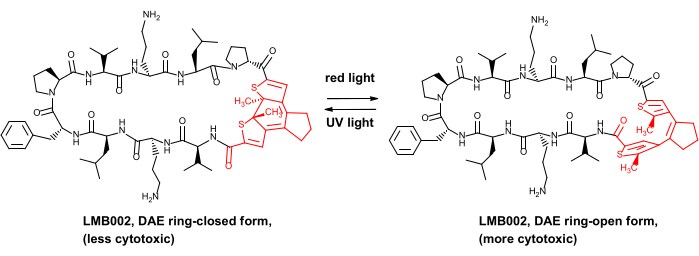
Figure 1 : Peptide cytotoxique photocontrôlé LMB002 et sa photoisomérisation. Le fragment de diaryléthène est représenté en rouge. Abréviation : DAE = diaryléthène. Veuillez cliquer ici pour voir une version agrandie de cette figure.
La recherche de résultats positifs et l’optimisation du hit-to-lead nécessitent souvent un criblage in vitro et in vivo de banques de composés appropriées 9,10. Nous démontrons ici une méthodologie adaptée au criblage systématique à haut débit de la cytotoxicité de composés photocontrôlés. Nous déterminons également l’efficacité de la photoisomérisation, estimons la dose lumineuse dans les tissus modèles et évaluons l’efficacité in vivo des candidats les plus performants. L’approche est conforme aux considérations de bioéthique et de soins aux animaux.
Dans ce travail, les méthodes précliniques traditionnelles sont modifiées pour éviter la photoisomérisation non contrôlée des composés testés. L’objectif global de l’application de ces méthodes modifiées ici est de développer une stratégie générale qui est simple et rapide et qui produit des données statistiquement significatives pour comparer de manière fiable les activités in vitro et rationaliser les tests d’efficacité in vivo des composés photocommutables pour l’identification et le développement ultérieur du plomb.
La stratégie comporte trois étapes consécutives. La première étape consiste à déterminer la CI 50 (viabilité cellulaire apparente à50 %) dans les dilutions en série pour les photoformes actives et inactives de composés biologiquement actifs photocontrôlés sélectionnés à l’aide de cultures cellulaires bidimensionnelles (2D, monocouche) et tridimensionnelles (3D, sphéroïdes) et de microscopie confocale automatisée à fluorescence à haut débit. Les fenêtres photothérapeutiques sont comparées par rapport à la différence IC50 entre les deux photoformes, et les candidats les plus performants sont sélectionnés. Il n’y a pas d’avantage particulier à évaluer la toxicité par microscopie automatisée et autres plateformes de dépistage de la cytotoxicité (essais)11; Des modèles tumoraux cellulaires plus complexes12 pourraient être facilement mis en œuvre à ce stade.
Pour les composés sélectionnés à l’étape 1, la deuxième étape consiste à estimer de manière réaliste leur efficacité de photocommutation à l’intérieur des tissus en fonction de la profondeur à partir de la surface du tissu irradié en quantifiant l’efficacité de photocommutation des photoformes moins actives dans un substitut tissulaire par chromatographie liquide à haute performance (CLHP) détectée par UV d’extraits d’échantillons irradiés. In vivo , l’efficacité de la photocommutation pourrait être étudiée, mais nous proposons d’utiliser un substitut tissulaire simple – la viande de porc hachée. Nous avons testé la validité de cette approche. Nous avons mesuré la conversion de nos composés photocommutables in vivo sur un modèle de cancer de souris et observé approximativement la même photoconversion à une profondeur mesurée lors d’expériences précédentes avec des souris8. Tout autre tissu artificielapproprié 13, tissu/organe bioimprimé en 3D14, matériel de biopsie ou autre matériel animal exempté pourrait être utilisé. Cependant, cette configuration est un bon compromis car elle est économique, rapide et éthique.
La troisième étape est la détermination de l’efficacité anticancéreuse in vivo dans un modèle de cancer murin. Les composés présentant des caractéristiques supérieures dans les expériences in vitro et une photocommutation efficace à une profondeur d’au moins 1-1,5 cm dans les tissus modèles sont sélectionnés pour cette expérience.
Ce protocole peut être appliqué à des composés possédant différents types de photocommutateurs, à condition que leurs photoformes (ou leurs états photostationnaires, PSS) soient stables pendant un temps raisonnable (quelques jours ou plus). À titre d’illustration, un LMB002 dérivé du DAE décrit précédemment est utilisé15. Les photoformes LMB002 sont thermiquement stables et peuvent être stockées à −20 °C pendant au moins un an sans dégradation substantielle. Les cellules de carcinome pulmonaire de Lewis (LLC) sont choisies pour cette démonstration in vitro et in vivo, mais aucune restriction n’est imposée sur le type cellulaire. Les cellules LLC sont adhérentes, facilement cultivables en 3D et utilisées pour générer des tumoroïdes (comme décrit dans la référence16). In vivo Les cellules LLC sont utilisées pour modéliser les processus métastatiques et peuvent facilement générer des tumeurs solides chez des souris immunocompétentes après injection sous-cutanée. Cette méthodologie in vivo peut être universellement appliquée à d’autres modèles de cancer17,18. La mise en œuvre détaillée de cette stratégie est décrite ci-dessous.
Protocol
Representative Results
Discussion
Les composés photocontrôlés sont sans précédent dans le développement de médicaments; cependant, aucune méthode n’a été établie pour leur évaluation préclinique et clinique. L’analogue de monothérapie le plus proche, la thérapie photodynamique (PDT), est la modalité de traitement à usage clinique adoptée par de nombreux pays contre le cancer et est en cours de développement pour d’autres indications19,20. Semblable à la photopharmacologie, la PDT est également basée sur l’utilisation de la lumière pour activer la substance bioactive (oxygène singulet). Par conséquent, certaines méthodes expérimentales utilisées pour les études précliniques et cliniques en PDT peuvent être adoptées pour la photopharmacologie. Par exemple, les sources lumineuses, les approches d’administration de la lumière et les dispositifs médicaux sont bien développés et approuvés pour la TPD; Ils peuvent être directement utilisés pour l’évaluation de médicaments photocontrôlés. Cependant, la PDT et la photopharmacologie présentent de nombreuses distinctions l’une del’autre4, ce qui justifie la nécessité d’établir des méthodes spécifiques pour cette dernière.
Premièrement, la substance non activée dans la PDT (oxygène) est toujours présente dans les tissus vivants à des concentrations non toxiques. En revanche, les composés biologiquement actifs photocontrôlés non activés peuvent avoir une activité résiduelle et une toxicité indésirable. Par conséquent, les médicaments de photopharmacologie idéaux devraient avoir minimisé l’activité biologique sous leur forme administrée et doivent être très actifs dans leur forme générée par la lumière, la « fenêtre photothérapeutique»21 doit être aussi grande que possible. Trouver le hit et effectuer l’optimisation hit-to-lead nécessite l’identification de composés appropriés et le criblage de bibliothèques relativement grandes, déjà aux premiers stades du développement de médicaments. Ici, nous avons proposé une microscopie fluorescente confocale automatisée à haut débit pour identifier les composés de photocommutation efficaces.
La méthode choisie d’évaluation de la cytotoxicité permet de mettre en œuvre facilement l’exigence la plus critique – le maintien du PSS ou la stabilité du photoisomère sensible à la lumière visible. En effet, lors de sa mise en œuvre, l’exposition à la lumière est minimisée. Par conséquent, si vous choisissez des méthodes alternatives, les méthodes automatisées devraient être préférées. Cette approche est fiable et informative. L’utilisation de cultures cellulaires 3D (sphéroïdes) à ce stade fournit une compréhension holistique de la réponse de la cellule au traitement dans un microenvironnement tissulaire plus réaliste. En outre, des informations précieuses sur le mécanisme d’action des composés peuvent être obtenues en utilisant la microscopie comme méthode directe. La microscopie confocale fluorescente avec protocole de coloration approprié permet l’évaluation visuelle de la morphologie des cellules et des sphéroïdes; Des détails importants sur la mort cellulaire et les changements à l’intérieur des cellules peuvent également être détectés.
Deuxièmement, l’application légère nécessite un choix judicieux du dosage de la lumière. Dans la PDT, le surdosage léger est extrêmement nocif pour les tissus22. La thérapie photopharmacologique peut être avantageuse en cas d’irradiation excessive de la lumière. La limite supérieure de la substance activée est définie par la dose administrée de la substance non activée et sa pharmacocinétique. Cependant, le dosage de la lumière est toujours un problème en photopharmacologie. Il faut veiller à ce que la densité de puissance d’irradiation et le temps d’exposition ne soient pas inférieurs à l’exigence du traitement. En principe, la génération de la substance activée peut être surveillée in vivo. Cependant, pour des raisons bioéthiques, nous avons proposé une expérience avec un tissu modèle (viande hachée fraîche) mélangé au composé non activé15. Cette expérience est simple et peut être modifiée pour utiliser différentes sources lumineuses. Il peut également être adapté pour l’estimation photophysique du dosage de la lumière et la mesure des influences thermiques. Là encore, en utilisant des tissus modèles, il est possible de minimiser l’exposition à la lumière, comparée, par exemple, à la détermination plus précise de l’efficacité de la photocommutation dans les conditions in vivo , une alternative qui peut toujours être intéressante à considérer.
Enfin, les composés qui présentent des caractéristiques supérieures dans les criblages de toxicité in vitro et qui photocommutent efficacement à au moins 1-1,5 cm de profondeur dans le tissu modèle peuvent être sélectionnés pour des études in vivo coûteuses, laborieuses et longues. Dans ce protocole, nous avons utilisé la même lignée cellulaire (LLC) que dans l’évaluation in vitro pour générer le modèle de cancer allogreffé. La dynamique de croissance tumorale, la mortalité et le nombre de métastases sont les paramètres les plus appropriés pour évaluer l’efficacité anticancéreuse. Par rapport à la chimiothérapie conventionnelle, un facteur supplémentaire est appliqué dans le traitement photopharmacologique – la lumière. Par conséquent, deux groupes d’animaux témoins sont nécessaires : l’un qui reçoit uniquement le véhicule et l’autre qui reçoit le véhicule et l’irradiation. Cette configuration permet d’évaluer l’impact de la lumière sur les paramètres mesurés. Dans notre expérience, les animaux des deux groupes expérimentaux ont reçu le composé non activé et les tumeurs des souris d’un groupe ont été irradiées. Le régime d’irradiation était identique pour les groupes témoin et de traitement. La comparaison avec la chimiothérapie de référence n’est pas nécessaire à ce stade car le but principal de l’expérience est de démontrer l’effet combiné de l’application de la lumière et du composé. Les composés les plus performants présentant cet effet peuvent ensuite être sélectionnés pour une étude plus approfondie de leur toxicité in vivo et une comparaison avec des critères de référence pour prendre des décisions importantes sur leur développement. Techniquement, l’expérience in vivo que nous décrivons peut être facilement adaptée à des études pharmacocinétiques ou pharmacodynamiques, par exemple, d’un composé déjà sélectionné comme médicament principal.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Les auteurs reconnaissent le financement de l’UE par le programme H2020-MSCA-RISE à travers les projets PELICO (#690973) et ALISE (#101007256). Ce travail a été soutenu par le DFG-GRK 2039 (SA, TS et ASU), le programme NACIP de la Société Helmholtz (SA et ASU) et le VIP+ du BMBF (OB et ASU). Nous remercions le Dr Serhii Koniev, de l’Institut de technologie de Karlsruhe, qui a synthétisé le composé LMB002, l’a purifié et a aimablement fourni le composé pour l’étude. Les auteurs remercient également Chupryna Maksym qui a filmé et compilé la vidéo en Ukraine, et tous les courageux défenseurs de l’Ukraine qui ont rendu possible le travail expérimental, l’écriture et le tournage de cette publication.
Materials
| Agilent 1100 Series capillary LC system | ALSI-Chrom (Agilent distributor) | – | |
| ATCC CRL-1642, LL/2 (LLC1) Lewis lung carcinoma cell line | ECACC | 90020104 | |
| C57BL/6NCrl mice, female, inbred | Charles River | Strain code: 027 | |
| CelCulture, CO2 incubator | Esco Micro | CCL-170B | |
| Corning Matrigel Basement membrane matrix | Merck | CLS354234 | |
| Corning, 384- well spheroid microplates | Merck | CLS3830 | |
| Fetal bovine serum | Merck | F7524 | |
| Gibco, DPBS | Thermo Fisher Scientific | 21600044 | |
| Gramicidin S | Lumobiotics | Custom synthesis | |
| HyClone, DMEM/high glucose | Cytiva | SH30003.04 | |
| IN Cell Analyzer 6500HS, imaging system | Cytiva | 29240358 | |
| Invitrogen, Calcein AM | Thermo Fisher Scientific | C1430 | |
| Isoflurane anesthesia machine | ASA | S/N ASA 1305 | |
| L-glutamine, 200 mM solution | Merck | G7513 | |
| LIKA-surgeon, diode surgery laser | Fotonika plus | – | |
| LMB002 | Lumobiotics | Custom synthesis | |
| Penicillin–Streptomycin, solution stabilized | Merck | P4333 | |
| PhenoPlate, 96-well plates | PerkinElmer | 6055302 | |
| Photometer PCE-LED 20 | PCE Instruments | PCE-LED 20 | |
| Thermo Scientific, Hoechst 33342 | Thermo Fisher Scientific | 62249 | |
| Thermo Scientific, Propidium iodide | Thermo Fisher Scientific | J66764-MC | |
| Trypan blue, 0.4% solution | Merck | T8154 | |
| Trypsin–EDTA, 10 x solution | Merck | T4174 | |
| UltraCruz Cell culture flasks with vented caps, 75 cm2 | Santa Cruz Biotechnology | sc-200263 | |
| UltraCruz, bottle top filters, PES, 0.22 μm | Santa Cruz Biotechnology | sc-360882 | |
| Vydac 218TP, C18 HPLC column (4.6 mm × 250 mm, 5 µm) | Altmann Analytik (Avantor distributor) | GR5103827 |
References
- Fuchter, M. J. On the promise of photopharmacology using photoswitches: a medicinal chemist’s perspective. Journal of Medicinal Chemistry. 63 (20), 11436-11447 (2020).
- Volarić, J., Szymanski, W., Simeth, N. A., Feringa, B. L. Molecular photoswitches in aqueous environments. Chemical Society Reviews. 50, 12377-12449 (2021).
- Paoletti, P., Ellis-Davies, G. C. R., Mourot, A. Optical control of neuronal ion channels and receptors. Nature Reviews Neuroscience. 20, 514-532 (2019).
- Hüll, K., Morstein, J., Trauner, D. In Vivo Photopharmacology. Chemical Reviews. 118 (21), 10710-10747 (2018).
- Ma, X., et al. In vivo photopharmacology with a caged mu opioid receptor agonist drives rapid changes in behavior. Nature Methods. 20, 682-685 (2023).
- Sarabando, S. N., Palmeira, A., Sousa, M. E., Faustino, M. A. F., Monteiro, C. J. P. Photomodulation Approaches to Overcome Antimicrobial Resistance. Pharmaceuticals. 16 (5), 682 (2023).
- Kolarski, D., Szymanski, W., Feringa, B. L., Hirota, T., Hatori, M., Panda, S. Chronophotopharmacology: Methodology for high spatiotemporal control over the circadian rhythm with light. Neuromethods. 186, (2022).
- Babii, O., et al. Peptide drugs for photopharmacology: how much of a safety advantage can be gained by photocontrol. Future Drug Discovery. 2 (1), FDD28 (2020).
- Davis, A. M., Keeling, D. J., Steele, J., Tomkinson, N. P., Tinker, A. C. Components of successful lead generation. Current Topics in Medicinal Chemistry. 5 (4), 421-439 (2005).
- Balani, S. K., Miwa, G. T., Gan, L., Wu, J., Lee, F. W. Strategy of utilizing in vitro and in vivo adme tools for lead optimization and drug candidate selection. Current Topics in Medicinal Chemistry. 5 (11), 1033-1038 (2005).
- Kleijn, A., et al. A Systematic comparison identifies an ATP-based viability assay as most suitable read-out for drug screening in glioma stem-like cells. Stem Cells International. 2016, (2016).
- Rodrigues, J., Heinrich, M. A., Teixeira, L. M., Prakash, J. 3D in vitro model revolution: unveiling tumor-stroma interactions. Trends in Cancer. 7 (3), 249-264 (2021).
- Sittinger, M., et al. Tissue engineering and autologous transplant formation: practical approaches with resorbable biomaterials and new cell culture techniques. Biomaterials. 17 (3), 237-242 (1996).
- Matai, I., Kaur, G., Seyedsalehi, A., McClinton, A., Laurencin, C. T. Progress in 3D bioprinting technology for tissue/organ regenerative engineering. Biomaterials. 226, 119536 (2020).
- Babii, O., et al. Direct photocontrol of peptidomimetics: an alternative to oxygen-dependent photodynamic cancer therapy. Angewandte Chemie International Edition. 55 (18), 5493-5496 (2016).
- De Ridder, K., et al. Novel 3D lung tumor spheroids for oncoimmunological assays. Advanced NanoBiomed Research. 2 (4), 2100124 (2022).
- Pauli, C., et al. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discovery. 7 (5), 462-477 (2017).
- Van Straten, D., Mashayekhi, V., De Bruijn, H. S., Oliveira, S., Robinson, D. J. Oncologic photodynamic therapy: basic principles, current clinical status and future directions. Cancers. 9 (2), 19 (2017).
- Li, X., Kwon, N., Guo, T., Liu, Z., Yoon, J. Innovative strategies for hypoxic-tumor photodynamic therapy. Angewandte Chemie International Edition. 57 (36), 11522-11531 (2018).
- Hull, K., Morstein, J., Trauner, D. In vivo photopharmacology. Chemical Reviews. 118 (21), 10710-10747 (2018).
- Babii, O., et al. Structure-activity relationships of photoswitchable diarylethene-based β-hairpin peptides as membranolytic antimicrobial and anticancer agents. Journal of Medicinal Chemistry. 61 (23), 10793-10813 (2018).
- Heckl, C., Aumiller, M., Rühm, A., Sroka, R., Stepp, H. Fluorescence and treatment light monitoring for interstitial photodynamic therapy. Photochemistry and Photobiology. 96 (2), 388-396 (2020).
