- 00:00Overview
- 01:22Principles of HPLC
- 05:05Preparation of the Mobile Phase
- 06:08Preparation of Component and Standard Solutions
- 07:25HPLC Setup
- 09:25Diet Soda Samples
- 10:16Results
- 11:27Applications
- 12:44Summary
고성능 액체 크로마토그래피 (HPLC)
English
Share
Overview
출처: 폴 바우어 박사 – 퍼듀 대학교
고성능 액체 크로마토그래피(HPLC)는 액체 샘플의 성분을 분리하고 정량화하는 데 일반적으로 사용되는 중요한 분석 방법입니다. 이 기술에서 솔루션(제1 상)은 표면에 결합된 두 번째 위상을 가진 작은 다공성 입자의 포장을 포함하는 열을 통해 펌핑된다. 두 단계에서 샘플 구성 요소의 용해도가 다르면 구성 요소가 서로 다른 평균 속도로 컬럼을 통과하여 이러한 구성 요소를 분리합니다. 펌핑된 솔루션을 이동단계라고 하며 컬럼의 위상을 고정 위상이라고 합니다.
액체 크로마토그래피의 여러 모드가 있습니다, 고정 및 / 또는 모바일 단계의 종류에 따라 고용. 이 실험은 고정 단계가 극적이지 않으며 이동 상이 극성인 반전 위상 크로마토그래피를 사용합니다. 사용되는 고정 단계는 C18 탄화수소 그룹이 3μm 실리카 입자에 접합되는 반면, 이동상은 극성 유기 수정자(acetonitrile)가 첨가된 수성 완충제로 용출 강도를 변화시킵니다. 이 양식에서 실리카는 수용성 샘플에 사용할 수 있어 광범위한 응용 을 제공합니다. 이 실험에서, 다이어트 청량 음료에서 자주 발견 되는 세 가지 구성 요소의 혼합물 (즉 카페인, 벤조아테, 그리고 아스파타메) 분리. 3종의 알려진 양을 포함하는 7개의 준비된 해결책이 이용되고, 그들의 크로마토그램은 그 때 기록됩니다.
Principles
HPLC 실험 중에 고압 펌프는 인젝터를 통해 저수지에서 이동 단계를 수행합니다. 그런 다음 부품 분리를 위해 역상 C18로 포장된 열을 통과합니다. 마지막으로, 이동상은 흡수도가 220nm에서 측정되고 폐병으로 끝나는 검출기 세포로 이동합니다. 구성 요소가 인젝터 포트에서 검출기로 이동하는 데 걸리는 시간을 보존 시간이라고 합니다.
이 실험에서는 역상 컬럼에서 분리가 수행되는 액체 크로마토그래프가 사용됩니다. 컬럼 치수는 3mm(즉, 100mm) x 100mm이며 실리카 패킹(3-μm 입자 크기)은 C18 옥타데실린실레인(ODS)으로 기능성된다. Rheodyne 6 포트 회전 사출 밸브는 처음에 작은 루프에 샘플을 저장하는 데 사용되며 밸브의 회전 시 이동 단계에 샘플을 소개합니다.
검출은 220 nm의 파장에서 흡수 분광법에 의한 것입니다. 이 실험은 검출기가 가변적이지 않은 경우 254nm에서 실행할 수 있습니다. 검출기의 데이터에는 디지털 멀티미터(DMM)를 사용하여 측정되고 데이터 수집 프로그램이 로드된 컴퓨터에서 읽는 아날로그 전압 출력이 있습니다. 생성된 크로마토그램은 샘플의 모든 구성 요소에 대한 피크를 가지고 있습니다. 이 실험을 위해, 세 가지 구성 요소는 모두 5 분 이내에 elute.
이 실험은 동위 동위 제일 상이라고 하는 단일 이동 단계 및 펌프를 사용합니다. 분리하기 어려운 샘플의 경우 그라데이션 모바일 단계를 사용할 수 있습니다. 이는 초기 이동 단계가 주로 수성 단계이며 시간이 지남에 따라 두 번째 유기 적인 모바일 단계가 점차 전체 모바일 단계에 추가되는 경우입니다. 이 방법은 시간이 지남에 따라이 단계의 극성을 제기하여 구성 요소의 보존 시간을 낮추고 가스 크로마토그래프의 온도 그라데이션과 유사하게 작동합니다. 컬럼이 가열되는 경우도 있습니다(일반적으로 40°C)는 주변 온도 변화와 관련된 보존 시간 오류를 없애는 경우가 있습니다.
역상 HPLC에서 컬럼 고정 위상 패킹은 일반적으로 C4, C8 또는 C18 패킹입니다. C4 컬럼은 주로 큰 분자량을 가진 단백질을 위한 반면, C18 컬럼은 펩타이드와 저분자량을 가진 기본 샘플을 위한 것입니다.
흡수 분광법에 의한 검출은 성분의 흡수 스펙트럼이 모두 쉽게 사용할 수 있기 때문에 압도적으로 선택의 검출 방법입니다. 일부 시스템은 전도도 또는 암페어와 같은 전기 화학 적 측정을 검출 방법으로 사용합니다.
이 실험을 위해, 이동상은 주로 20% 아세토닐릴과 80% 정제된 디온화(DI) 물이다. 소량의 아세트산이 첨가되어 이동상pH를 낮추어 고정된 패킹 단계에서 실라놀을 유지하여 비소화 상태로 유지합니다. 이렇게 하면 흡착 피크가 테일링에서 감소하여 더 좁은 피크를 제공합니다. 이어서, pH는 40% 수산화나트륨으로 조정되어 pH를 높이고 성분의 보유 시간을 줄이는 데 도움을 줍니다.
각 그룹은 표준 용액의 상이한 농도를 포함하는 7 개의 바이알 세트를 사용합니다(표 1). 처음 3은 각 피크를 식별하는 데 사용되며 마지막 4는 각 구성 요소에 대한 교정 차트를 만드는 것입니다. 표준 1-3은 교정 차트에도 사용됩니다.
| 수 | 카페인 (mL) | 벤조아테 (mL) | 아스파타메 (mL) |
| 1 | 4 | 0 | 0 |
| 2 | 0 | 4 | 0 |
| 3 | 0 | 0 | 4 |
| 4 | 1 | 1 | 1 |
| 5 | 2 | 2 | 2 |
| 6 | 3 | 3 | 3 |
| 7 | 5 | 5 | 5 |
표 1. 7개의 제공된 작업 표준을 준비하는 데 사용되는 재고 표준의 볼륨(각 표준의 총 부피는 50mL).
Procedure
Results
The HPLC chromatograms are able to quantify each of the 3 components for all the samples based upon the calibration curves of the standards (Figure 3).
From this set of experiments, it was determined that a 12-oz can of these diet sodas contained the following amounts of each component:
Diet Coke: 50.5 mg caffeine; 217.6 mg aspartame; 83.6 mg benzoate.
Coke Zero: 43.1 mg caffeine; 124.9 mg aspartame; 85.3 mg benzoate.
Diet Pepsi: 34.1 mg caffeine; 184.7 mg aspartame; 79.5 mg benzoate.
Not surprisingly, all 3 had roughly the same amount of benzoate, as it is just a preservative. The Coke products had a bit more caffeine, and the Coke Zero had much less aspartame than the other two sodas, as it also includes citric acid for some flavoring.
The following numbers are the actual amounts of caffeine and aspartame in a 12-oz can of the 3 diet sodas (The caffeine content was obtained from the Coca-Cola and Pepsi websites. The aspartame content was obtained from both LiveStrong.com and DiabetesSelfManagement.com.):
Diet Coke: 46 mg caffeine; 187.5 mg aspartame
Coke Zero: 34 mg caffeine; 87.0 mg aspartame
Diet Pepsi: 35 mg caffeine; 177.0 mg aspartame
Sample Calculations (Table 2):
Concentration of caffeine in STD#1: The component solution for caffeine had 0.400 g of caffeine diluted to 500 mL = 0.500 L → 0.800 g/L = 0.800 mg/mL.
STD#1 had 1 mL of this solution diluted to 50.0 mL
0.800 mg/mL * (1.0 mL / 50.0 mL) = 0.016 mg/mL = 16.0 mg/L.
STD#2 had 2 mL of this solution diluted to 50.0 mL
0.800 mg/mL * (2.0 mL / 50.0 mL) = 0.032 mg/mL = 32.0 mg/L.
The results from the three calibration graphs (Figure 4) yielded the following equations:
Caffeine Peak Area = 0.1583*[Caffeine mg/L] – 0.574
Aspartame Peak Area = 0.02696*[Aspartame mg/L] – 0.405
Benzoate Peak Area = 0.1363*[Benzoate mg/L] – 1.192
Diet Coke: Caffeine Peak Area = 10.68 = 0.1583*[Caffeine mg/L] – 0.574
[Caffeine mg/L] = (10.68 + 0.574)/ (0.1583) = 71.1 mg/L in the injected sample.
Since the sample was diluted by a factor of 2, the Diet Coke had 141.2 mg/L caffeine.
The amount per 12-oz can = (141.2 mg/L)(0.3549 mL/12-oz can) = 50.5 mg caffeine/can.
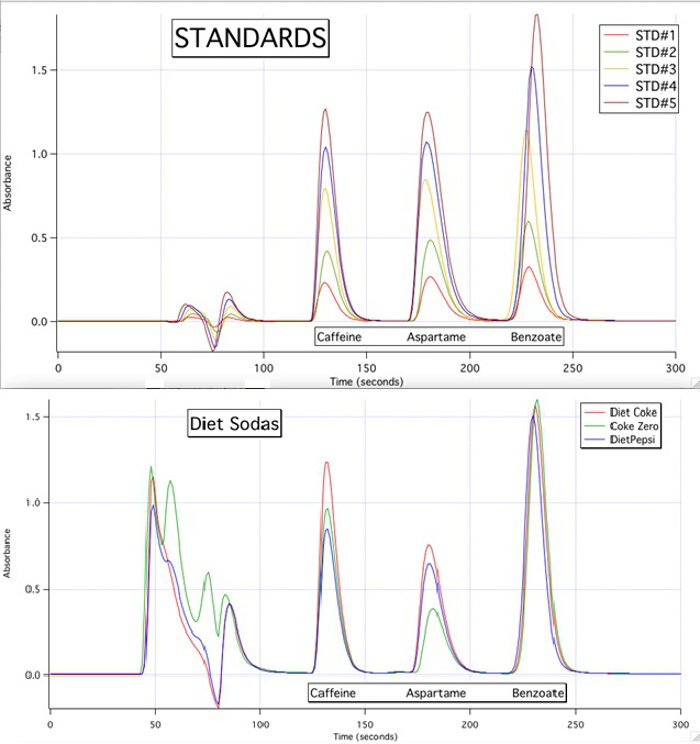
Figure 3. The HPLC chromatograms of the 5 standards and the 3 samples.
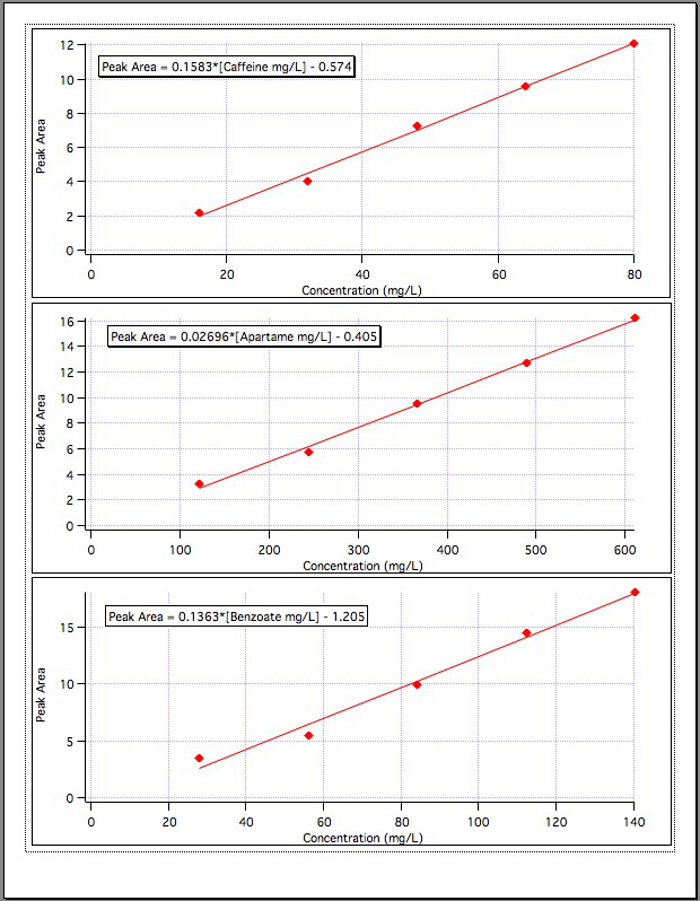
Figure 4. The calibration curves for each of the 3 components.
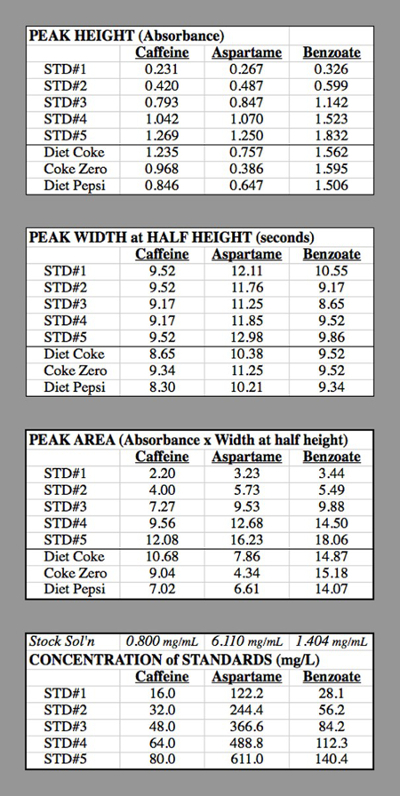
Table 2. The data tables for the HPLC trials used for generating the calibration curves.
Applications and Summary
HPLC is a widely-used technique in the separation and detection for many applications. It is ideal for non-volatile compounds, as gas chromatography (GC) requires that the samples are in their gas phase. Non-volatile compounds include sugars, vitamins, drugs, and metabolites. Also, it is non-destructive, which allows each component to be collected for further analysis (such as mass spectrometry). The mobile phases are practically unlimited, which allows changes to the polarity of pH to achieve better resolution. The use of gradient mobile phases allows for these changes during the actual trials.
There has been concern over the possible health issues that may be associated with the artificial sweetener aspartame. Current product labeling does not show the amount of these components inside of the diet beverages. This method allows for quantifying these amounts, along with the caffeine and benzoate.
Other applications include determining the amounts of pesticides in water; determining the amount of acetaminophen or ibuprofen in pain reliever tablets; determining whether there are performance-enhancing drugs present in the bloodstream of athletes; or simply determining the presence of drugs in a crime lab. While the concentrations of these samples, and often the identity of the components, can be readily determined, the one limitation is that several samples could have close to identical retention times, resulting in co-eluting.
Transcript
High-performance liquid chromatography, or HPLC, is a highly versatile technique that separates components of a liquid mixture based on their different interactions with a stationary phase.
HPLC is an adaptation of column chromatography. In column chromatography, a column is packed with micro-scale beads called the stationary phase. The stationary phase beads are functionalized with chemical groups that induce an interaction between the bead and the components of a mixture located in the liquid, or mobile phase. As the mixture flows through the column, the components interact with the stationary phase differently.
In HPLC, column chromatography is performed at a higher flow rate, and therefore higher pressure, than classical column chromatography. This enables the use of smaller stationary phase beads with a greater surface area to volume ratio, which greatly increases the interaction of the stationary phase and components in the mobile phase.
This video will introduce the basics of the operation of HPLC by demonstrating the separation of components of various diet sodas.
There are two types of HPLC used in the laboratory: analytical, and preparative. In analytical HPLC, the instrument is used to identify components of a small volume, and the analyzed sample is then discarded as waste. In preparative HPLC, the instrument is used to purify a mixture and a desired amount of each component is collected in fractions.
The HPLC instrumentation consists of a series of simple components. First, the mobile phase, held in solvent reservoirs, is pumped through the system by one or more pumps at a constant flow rate. The sample is injected into the mobile phase stream by the sample injector. The sample, diluted by the mobile phase, is then delivered to the HPLC column, where the components of the sample are separated. The components are then analyzed by the detector, and either saved in fractions for later use, or transferred to a waste bottle.
The HPLC column is the key component to the system. It is composed of a metal or plastic cylinder, packed with micro-scale beads of stationary phase, or chromatography resin. The sample mixture flows through the packed particle bed at a constant flow rate and each component interacts with the stationary phase as it flows by.
The compounds interact with the stationary phase differently, and therefore travels down the length of the column to the detector at a different rate. The time required for a component to exit the column, or elute, is called the retention time. The result is a plot of retention time vs. intensity, or a chromatogram. The retention time is used to identify the component. The peak size, specifically the area under the peak, is used to quantify the amount of the compound in the initial solution.
The choice of stationary phase depends on the properties of the components in the sample mixture. The most commonly used stationary phase is silica beads, as they are an inert nonpolar material that forms micro-scale beads, and achieves sufficient packing density. The most common type of HPLC is reversed-phase chromatography, which utilizes a hydrophobic stationary phase, typically silica beads with C18 chains bonded to the beads’ surface. The components are eluted in order of decreasing polarity.
The mobile phase used in reversed-phase chromatography is typically a mixture of water and an organic solvent, such as acetonitrile. Depending on the sample, the mobile phase can remain a constant ratio of water and organic solvent, known as isocratic mode. However, this can lead to broad peaks, in the case of high water content, or overlapping peaks—in the case of high organic content.
The mobile phase ratio can also be changed linearly or stepwise during the separation, to create a mobile phase gradient. A gradient elution can prevent peak broadening of the less polar components, thereby improving the separation and shortening the elution time.
Now that the basics of HPLC have been outlined, the HPLC technique will be demonstrated in the laboratory. In this experiment, HPLC will be used to separate and quantify three common components of diet soda.
First, to prepare the mobile phase, add 400 mL of acetonitrile to 1.5 L of purified deionized water. Then carefully add 2.4 mL of glacial acetic acid. Dilute the solution to a total volume of 2 L. The resulting solution should have a pH between 2.8 and 3.2.
Adjust the pH to 4.2 by adding 40% NaOH, drop-wise to the stirring solution, with the use of a calibrated pH meter.
Filter the mobile phase through a 0.47-μm membrane filter under vacuum to degas the solution and remove solids that could plug the column. It is important to degas the solution, as bubbles can cause voids in the stationary phase, or work their way to the detector cell and cause instability in measurements.
Prepare three component solutions of caffeine, benzoate, and aspartame, which are three typical components of diet sodas. These component solutions are then used to prepare the standard solutions that will be utilized to determine the unknowns. Prepare 500 mL of the caffeine and benzoate solutions.
Prepare 100 mL of the aspartame component solution. Store the solution in the refrigerator when not in use to avoid decomposition.
Next, prepare 7 standard solutions, each with different concentrations of caffeine, benzoate, and aspartame. Pipet the proper amount of each component into a volumetric flask, and dilute to the 50-mL mark with mobile phase.
The first 3 solutions each contain one component, to enable peak identification. The other 4 solutions contain a range of concentrations of all 3 components, in order to correlate peak height to concentration.
Pour each standard solution into a labeled vial in a sample rack. Store the sample rack with samples and the remaining solutions in the refrigerator.
First, set up the mobile phase and waste containers. Ensure that the waste lines are fed into a waste container, and are not recycling back into the mobile phase. Ensure that the inlet mobile phase line is fed into the mobile phase container.
Verify that the flow rate of the mobile phase is set to 0.5 mL/min. This flow rate will enable all components to elute within 5 min, but is slow enough to ensure resolution of individual peaks.
Next, verify the minimum and maximum pressures on the solvent delivery system. These settings shut the pump off in case of a leak or clog, respectively.
Press “zero” on the detectors front panel, to set the blank. Rinse a 100-μL syringe with deionized water, then with several volumes of 1 of the 7 working standards. Then fill the syringe with that solution. Begin with the 3 single-component samples in order to identify the peak of each component.
Next, manually inject the solution, by placing the injector handle in the load position. Slowly inject the 100 μL of solution through the septum port.
Verify that the data collection program is set to collect data for 300 s, which allows for enough time for all 3 peaks to elute through the detector. When ready to begin the trial, rotate the injector handle to the inject position, in order to inject the sample into the mobile phase. Immediately, click “Start Trial” on the data collection program. When the scan is complete, repeat the process for each of the 7 standard solutions. For each of the first 3 standards, only one of the 3 peaks appears. Note the location of the peak, which is used to identify the component.
Select 3 diet soda samples, and allow them to sit out in open containers overnight to remove the carbonation.
After overnight degassing, draw approximately 3 mL of each diet soda into a plastic syringe. Next, attach a filter tip to the syringe and push the soda through the filter into a glass vial, in order to remove any solid particulates.
Dilute 2 mL of each sample with 2 mL of the mobile phase to decrease the soda concentration by half.
Draw 100 μL of one of the soda samples into a syringe, and inject it into the sample loop. Run the trial with identical parameters to the standard solutions. Repeat for each soda sample.
First, correlate the peak areas of the standard samples to the known concentrations. To do so, determine the peak areas on the chromatographs for each standard sample using the triangular method. Calculate the peak height times with the width at half of the height, and use this value as the peak area.
Using the peak area and known concentrations create a calibration curve for each component, and determine the least-squares fit for each calibration curve.
Calculate the concentration of each component in the diet sodas from the peak areas. Remember that the sodas were all diluted by a factor of 2 prior to injection into the HPLC. Based on these results, calculate the mg of each component in a 12-oz can of soda.
Unsurprisingly, all 3 sodas tested contained roughly the same amount of the preservative benzoate. However, the Coke products contained more caffeine. The calculated values for all components correlated well to reported values by the manufacturers.
HPLC is a highly versatile instrument, which is used in a wide range of analyses.
HPLC is often used to purify peptide molecules. In this example, transmembrane peptide complexes were prepared, and then stabilized by oxidative crosslinking the proteins with disulfide bonds.
The proteins were then dissolved in formic acid, and purified using reversed phase HPLC. The sample was then eluted using a linear gradient of two solvents, and the purity confirmed with mass spectrometry.
HPLC can also be used to identify organic compounds synthesized in the lab. In the Miller-Urey experiment, the abiotic synthesis of organic compounds on primordial earth was studied. Primordial gases, such as methane and ammonia, were introduced to a flask containing water, simulating early oceans. Electrical discharge was then applied, imitating lightning on primordial earth.
The water was then analyzed using HPLC coupled with mass spectrometry, and compared to known amino acid standards. 23 amino acids were synthesized and identified in this experiment.
You’ve just watched JoVE’s introduction to HPLC. You should now understand the basics of running the instrument, and analyzing the resultant data.
Thanks for watching!