Use of Fimbrial Rod for F18ab Fimbriae+ STEC Colonization to Host Cells
Summary
Here we present a protocol to study the function of fimbriae in bacterial colonization.
Abstract
Type 1 fimbriae are important virulence determinants of some Gram-negative pathogens, which promote bacterial colonization. The fimbrial rod is primarily composed of multiple copies of the major fimbrial subunit FimA. FimH adhesin, however, is present as a fibrillar tip structure that drive bacteria binding to host cellular mannose containing receptor. Here, we provide protocols to evaluate and compare the function of type 1 fimbrial subunits in F18ab fimbriae+ Shiga toxin-producing Escherichia coli (STEC). We found that both FimA and FimH are required for bacterial adhesion, invasion, and biofilm formation. Deleting fimA gene showed much more reduction in bacterial adhesion and invasion to porcine intestinal columnar epithelial cells IPEC-J2, than that of fimH mutant. Biofilm formation was significantly reduced in both mutants with an equal level. In addition, qPCR demonstrated that either fimA or fimH deletion down-regulated the bacterial flagella and F18 fimbriae genes expression, while up-regulated adhesin was involved in diffuse adherence-I (AIDA-I) gene expression, suggesting the co-regulation of cell surface-localized adhesins in F18ab fimbriae+ STEC.
Introduction
Bacterial fimbriae mediated adhesion facilitates bacterial attachment to a target cell surface and establishes an initial infection. Type 1 fimbriae are widely distributed among Escherichia coli (E. coli) and promote bacterial attachment to mammalian cells by binding to the mannose-containing receptor1,2,3. In contrast to pathogenic strains, 85% of tested commensal E. coli strains of human origin do not express type 1 fimbriae4, which indicates its critical roles in disease infection. Type 1 fimbriae are also important virulence factors for extra-intestinal pathogens, such as uropathogenic E. coli (UPEC) and neonatal meningitis-causing E. coli (NMEC)2,5,6.
Infections caused by F18 fimbriae+ (including two variants: ab and ac) Shiga toxin-producing E. coli (STEC) strains are associated with porcine edema disease (ED) and post-weaning diarrhea (PWD)7. Porcine F18 fimbriae+ STEC attaches to intestinal epithelial receptors by a variety of surface adhesins, including F18 fimbriae, flagella, E. coli common pilus (ECP) and the adhesin involved in diffuse adherence (AIDA-I)8,9,10,11. Previously, we had investigated the function of type 1 fimbriae in F18ac fimbriae+ ETEC, which demonstrated that type 1 fimbriae facilitate bacterial biofilm formation and adhesion to host cells12. However, as the pathogenesis of F18ab and F18ac fimbriae+ STEC are not totally the same7, the role of type 1 fimbriae in F18ab fimbriae+ STEC remains unclear. The fimbrial rod is primarily composed of multiple copies of the major fimbrial subunit FimA, and FimH adhesin is assembled into a fibrillar tip structure that drive bacteria binding to host cellular mannose containing receptor13. Using λ-Red recombination14, we had successfully knocked out fimA/fimH gene from a F18ab fimbriae+ STEC strain F107/86 (wild-type, O139:H1, Stx2e+), and constructed complement strains for this study15.
Here, we describe a protocol to study the function of bacterial fimbriae in colonization. Bacteria adhesion assay and invasion assay are major methods to investigate the bacteria fimbrial binding performance. It is complicated and costly to perform an animal challenge model or isolate the primary cell line for further infection assays16. Usually, neither of these results are stable with good repeatability since the individual differences are present between the tested animal. In this study, IPEC-J2 cells are used. These are porcine intestinal columnar epithelial cells that have been isolated from a neonatal piglet’s mid-jejunum17. It is a stable in vitro cell model for examining the interactions of various animal and human pathogens, including Salmonella enterica and pathogenic E. coli, with intestinal epithelial cells18, helping explain the role of fimbriae in intestinal infection conveniently and quickly. Otherwise, IPEC-1 cells are another widely used porcine intestinal epithelial cell line, in which case the composition of cellular receptors are different from IPEC-J219. For the study of mammary pathogenic bacteria, it is better to use mammary epithelial cell line MAC-T20. Hence, for different bacterial pathogenic conditions, choice of a suitable cell line which mimic in vivo environments is important.
In addition, the biofilm is another essential characteristic for bacterial survival during colonization21. In the previous works, silver and congo red were used to stain the biofilm formation in the glass tubes that visually showed the results22,23. However, the difference of biofilm formation ability between varying strains cannot be measured. Here, we also present a protocol for the quantification of bacterial biofilm formation in vitro, which could easily evaluate the ability of fimbriae in biofilm formation.
The methods proposed in this study utilize a fast and simple in vitro way to determine the function of bacterial fimbriae during the bacteria infection process, which can be widely adapted to other researches in the study of virulence factor in bacterial pathogenic mechanism.
Protocol
1. Cell culture
- Maintain IPEC-J2 cells in a 25 cm2 flask containing 5 mL of antibiotic-free F12-RPMI1640 (1:1) mixed media supplemented with 10% fetal bovine serum (FBS) at 37 °C, in a 5% CO2 incubator.
- One day before the adhesion assay, use 1 mL of 0.05% trypsin-EDTA solution to trypsinize IPEC-J2 cells for 3 min. Gently remove the trypsin-EDTA solution before cells start shedding from the flask. Add 3 mL of growth media and suspend the cells.
- Use 10 µL of the cell suspension to count the cells using a hemocytometer. Dilute the cell suspension using cell culture medium to a final concentration of 7 x 105 cells/mL in a 15 mL conical tube.
- Transfer 100 µL of cell suspension (~7 x 104 cells) to each well of a 96 well plate. Make sure that the cells distribute uniformly in the wells. Incubate the plate at 37 °C with 5% CO2 and allow the cells to adhere and grow overnight, which should be at about 90% confluency.
2. Bacteria adhesion and invasion assay
- Two days before the adhesion assay, streak frozen stocks of E. coli F107/86, ΔfimA mutant, ΔfimH mutant, ΔfimA/pfimA, and ΔfimH/pfimH onto separate LB agar plates to produce single colonies. Keep the plates in an incubator set to 37 °C to let the colonies grow overnight.
- One day before infection, pick a single colony from the bacterial culture plate using a sterile inoculation loop, and inoculate bacteria with 4 mL of Luria–Bertani (LB) broth in a bacterial glass culture tube. Cap the tubes after inoculating and put them into a shaker with 180 rpm at 37 °C, overnight.
- On the day of infection, take out bacterial cultures which have been growing overnight in the incubator. Transfer 30 µL of this culture (1:100 dilution) to a tube containing freshly prepared media. Place the tubes at 37 °C in a shaking incubator for 4 h (OD600 ~ 2.0, bacteria grown at mid-log phase).
- After 4 h, prepare 1 mL of sterile LB broth as a blank sample. Mix 100 µL of bacterial subculture and 900 µL of LB into one cuvette as a bacterial sample. Prepare the tubes for different bacterial samples.
- Measure the optical density (OD) of the bacterial cultures using a spectrophotometer. Measure OD at the wavelength of 600 nm (OD600). Measure the blank sample to get the background absorbance.
- Measure OD values for all the samples and record the OD600 values. Calculate the concentration accounting for the dilution factor (10 in this example).
- Dilute the culture with fresh F12-RPMI1640 (without FBS) to obtain an OD of approximately 0.1, which roughly corresponds with 1 x 108 cfu/mL. These bacterial suspensions will be later used as inoculum.
- Take out the 96 well plate containing the overnight cultured cells from the incubator. At this point, there are approximately 1 x 105 cells/well in the plate. Label the lid of the plate with the bacterial strain to be used for the infection of each well, with each infection being performed in triplicate.
- Remove the media from each well and gently wash each well with PBS three times to remove the non-adherent bacteria. Add 100 µL of the inoculum to the appropriate wells. Transfer the infected cells in an incubator maintained at 37 °C with 5% CO2.
- For bacteria adhesion assay, incubate infected cells in a 5% CO2 incubator at 37 °C for 1 h and directly move to step 2.12.
- For bacteria invasion assay, incubate infected cells in a 5% CO2 incubator at 37 °C for 2 h and go forward to step 2.10.
- After 2 h of incubation, take out the 96 well plate and aspirate the culture media with a pipette. Gently wash each well with PBS three times to remove the non-adherent bacteria.
- Add 200 µL of cell media with 100 μg/mL gentamicin to each well, incubate in a 5% CO2 incubator at 37 °C for 1 h to kill extracellular bacteria.
- After 1 h of incubation, take out the 96 well plate and remove the culture media. Gently wash each well with PBS three times to remove the non-adherent bacteria.
- Add 200 µL of 0.5% Triton X-100 to each well to lyse the cells and incubate for 20 min at room temperature.
- Transfer 200 µL of lysed cell from each well to a new 1.5 mL microcentrifuge sterile tube. Wash each well with 300 µL PBS and add the wash buffer to the 1.5 mL tube as well.
- Perform a 10-fold serial dilution of the collected lytic suspension with PBS in tubes.
- Plate 100 µL of the two lowest dilutions to the LB agar plate using a cell spreader to obtain single colonies. Incubate these plates overnight at 37 °C in an incubator.
- The following day count the colonies forming units, which represent the adherent / invasive bacteria number. Data is presented relative to the number of the WT strain, which was normalized to 100%. Each experiment needs to be repeated independently at least triplicate.
3. Biofilm formation quantification assay
- Two days before biofilm formation assay, prepare the bacteria strains as in step 2.1. Bacterial cultural plates were incubated at 37 °C overnight.
- One day before the assay, add 4 mL of biofilm-inducing media (for media composition please see ref.10) to sterile bacterial culture tubes. Pick a single colony from streaked bacterial cultures using a sterile inoculation loop and transfer the colony to the biofilm-inducing media. Cap the tubes after inoculating and put them into a shaker with 120 rpm at 30 °C, overnight.
- On the day of the assay, prepare the 96 well plate with a round bottom. Label the lid of the plate according to the incubated strain that will be used for each well, with each strain being done in triplicate.
- Transfer 10 µL of each overnight bacterial culture (1 in 100 dilution) to 990 µL fresh biofilm-inducing media in 1.5 mL microcentrifuge tubes. These bacterial suspensions will be later used as inoculum.
- Add 200 µL of different inoculum to appropriate wells of the plate in triplicate. Transfer the 96-well plate to an incubator at 37 °C for 24 h.
- After 24 h of incubation, take out the 96 well plate and remove the cultural media. Gently wash each well with double distilled water (ddH2O) three times to remove the uncombined bacteria.
- Add 250 µL of 2% crystal violet solution10 to each well and incubate at room temperature for 15 min to stain the biofilm.
- Remove the 2% crystal violet solution from the 96 well plate. Gently wash each well with ddH2O three times to remove the redundant dye. Then, transfer the plate to an incubator at 37 °C for 15 min to dry the wells.
- Add 300 µL of 95% ethanol to each well; solubilize the crystal violet stained on the bacterial biofilm.
- Turn on the 96 well spectrophotometer; set the detect absorbance as 600 nm. Put the 96 well plate on the load and start the detection.
- Compare the mean value of various samples. Data are presented relative to the absorbance of the WT strain, which was normalized to 100%. Each experiment needs to be repeated, independently, at least three times.
4. RNA isolation and reverse transcription
- Two days before the qPCR assay, streak frozen stocks of E. coli F107/86, ΔfimA mutant, ΔfimH mutant, ΔfimA/pfimA, and ΔfimH/pfimH onto the LB agar plates to produce single colonies, respectively. Transfer the plates to an incubator set to 37 °C and allow overnight growth.
- One day before the assay, add 4 mL of biofilm-inducing media to sterile bacterial culture tubes. Pick a single colony from streaked bacterial cultures using a sterile inoculation loop and touch the loop to the biofilm-inducing media. Cap the tubes after inoculating and put them into a shaker with 180 rpm at 37 °C, overnight.
- Transfer 30 µL of the overnight bacterial culture to 3 mL fresh LB media in the glass tubes on the day of the assay. Place the tubes into a shaking incubator at 37 °C with 180 rpm.
- After a 4 h culture (OD600 ~ 2.0, bacteria grown at mid-log phase), 1 mL of bacteria culture was collected by centrifugation (12,000 x g, 2 min) in a 2 mL sterile RNase-free microcentrifuge tube.
- Add 200 µL of lysozyme solution (1 mg/mL) into the 2 mL tube and incubate at 37 °C for 10 min.
- Add 800 µL of commercially available guanidium hydrochloride reagent to the tube; then, transfer the tube onto ice.
- Add 200 µL of chloroform to each sample tube. Then, carefully cap the tube and vortex for 15 s. At last, incubate the tube at room temperature for 10 min.
- The tabletop centrifuge is set to a temperature of 4 °C and pre-cold before use.
- Perform the centrifugation at 12,000 x g for 10 min at 4 °C.
- Prepare and label the new 1.5 mL RNase-free tubes. Transfer the upper aqueous phase to the new tube, being careful not to disturb the middle or lower layers.
- Add an equal volume of isopropanol into the tube; vortex the mixture of aqueous layer and isopropanol and allow samples to sit at room temperature for 10 min.
- Perform the centrifugation at 12,000 x g for 10 min at 4 °C.
- Watch the bottom of the tube to find whether there is a white pellet (RNA). Carefully pour out the supernatant from the tube into a waste container.
- Add 500 µL of 75% ethanol and vortex, to wash the RNA pellet.
- Perform the centrifugation once again, at 12,000 x g for 5 min at 4 °C.
- Carefully aspirate to remove the supernatant as much as possible.
- Air-dry the white RNA pellets in the benchtop until it turns clear.
- Transfer 30 µL of pre-cold RNase-free ddH2O to dissolve the RNA. Pass the solution a few times to help dissolve. Keep the samples on ice all along.
- Detect the RNA concentration of each sample using a micro spectrophotometer with 1 µL of the sample. Record the concentration of all the samples.
- To perform reverse transcription, prepare the following mixture in an RNase-free centrifuge tube: 4 μL of 4x master mix, 1 µg template RNA, and RNase-free ddH2O up to 16 μL.
- Mix gently with a pipette. Cap the samples tightly and label the PCR tubes on the side.
- Centrifuge the PCR tubes briefly (short spin) to collect the samples at the bottom of the tube.
- Place the PCR tubes in a thermocycler and run the samples under the following settings: 42 °C for 2 min and then hold at 4 °C.
- Add 4 μL of 5x Enzyme Mix to the mixture of the previous step; mix gently with a pipette.
- Briefly centrifuge the PCR tubes to collect samples to the bottom of the tube.
- Place the PCR tubes in a thermocycler and run the samples under the following settings: 37 °C for 15 min, 85 °C for 5 s, and then hold at 4 °C.
- Serially dilute the product 1:5 with ddH2O in the tubes, which can be directly used in qPCR reactions, or store at -20 °C for further use.
5. qPCR analysis
- Plan the setup of the 96 well qPCR plate for sample analysis.
- Thaw primers (for sequence see Table 1), master mixes, and cDNA on ice.
- Prepare the mix as follows for each well reaction: 10 μL of 2x SYBR qPCR Master Mix, 0.4 μL of Primer Forward and Primer Reverse, and ddH2O up to 18 μL.
NOTE: Major bacterial fimbriae/adhesin gene fragments, including fedF (encoding the adhesin of F18 fimbriae), fliC (encoding the flagellin), ecpA (encoding the major subunit of E. coli common pilus) and AIDA-I (encoding the adhesin involved in diffuse adherence) are amplified as target genes. gapA (encoding the glyceraldehyde 3-phosphate) is used as the reference gene. - Vortex the mix and centrifuge at 500 x g for 1 min. Using a repeater pipette, carefully transfer 18 µL of the mix into each well of the 96 well plate.
- Transfer 2 µL of diluted cDNA (from step 3.8) to triplicate wells for each primer set.
- Use an adhesive film to seal the plate surface, ensuring that all wells are covered. Use a roller to seal firmly.
- Centrifuge the plate at 500 x g for 1 min. An empty plate is used as a counterbalance.
- Turn on the Real-Time PCR System; follow the qPCR reagent instructions to set the parameters. Ensure that the Melt Curve is included in the program.
- Place the 96 well plate in the thermocycler and start the analysis.
- Compare the value of the various samples and analyze the data with the 2-ΔΔCT method24.
Representative Results
FimA is more important than FimH in F18ab fimbriae+ STEC adhesion and invasion to IPEC-J2 cells. Compared to WT strain, deleting fimA reduced F18ab fimbriae+ STEC adhesion to IPEC-J2 cells by approximately 86% (p < 0.01), while deleting fimH reduced STEC adhesion by approximately 71% (p < 0.01) (Figure 1A). Blocking the adhesin FimH of WT strain by co-incubating with 4% D-mannose showed an equal adhesion ability with the ΔfimH mutant, while the F107/86ΔfimA/pfimA and F107/86ΔfimH/pfimH restored bacterial adhesion to the same levels as the WT.
Likely, ΔfimA mutant only showed 36% of the ability of ΔfimH mutant in F18ab fimbriae+ STEC invasion to IPEC-J2 cells (p < 0.05) (Figure 1B). Both complemented strains were able to restore the invasion ability of the WT level.
Type 1 fimbriae contribute to biofilm formation in F18ab fimbriae+ STEC. The F107/86ΔfimA strain exhibited 17% of the WT strain absorbance of OD600 nm (Figure 2, p < 0.01), while ΔfimH exhibited 16% of the WT strain absorbance in the CV assay for biofilm formation (Figure 2). The biofilm formation capacity is not a significant difference between these two mutants.
Type 1 fimbriae deficiency affects the expression of other adhesins. Fimbriae and flagella are major bacterial surface structures that mediate bacteria-host interaction. Co-regulation of these cell surface-localized adhesins were found using qPCR (Figure 3). Deleting fimA reduced fliC (flagellin) and fedF (adhesive subunit of F18 fimbriae) expression to 73% and 71% (p < 0.05) compared to the WT levels, respectively. Similarly, when compared to the WT, fliC and fedF expression in fimH mutant reduced to 68% and 70% (p < 0.05), respectively.
By contrast AIDA-I expression in fimA and fimH mutant was respectively elevated 3.3- and 3.5-fold (p < 0.05), while ecpA expression was changed in neither mutant.
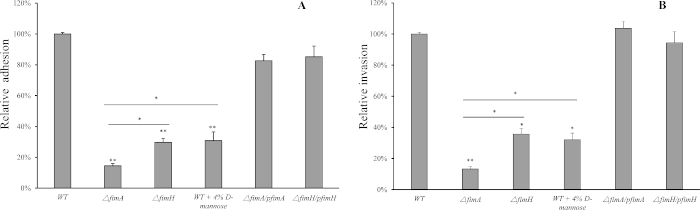
Figure 1: Both FimA and FimH subunits are required for F18ab fimbriae+ STEC adhesion and invasion to IPEC-J2 cells. (A). Wild type F18ab fimbriae+ STEC and the ΔfimA and ΔfimH mutants’ adherence to IPEC-J2 cells. (B). Wild type F18ab fimbriae+ STEC, the ΔfimA and ΔfimH mutants’ invasion to IPEC-J2 cells. Data is presented relative to the invasion of the WT strains to cells, which was normalized to 1.0. Mean and standard deviation of triplicate experiments are shown. Significant differences between different groups are indicated (* p < 0.05, ** p < 0.01). Please click here to view a larger version of this figure.
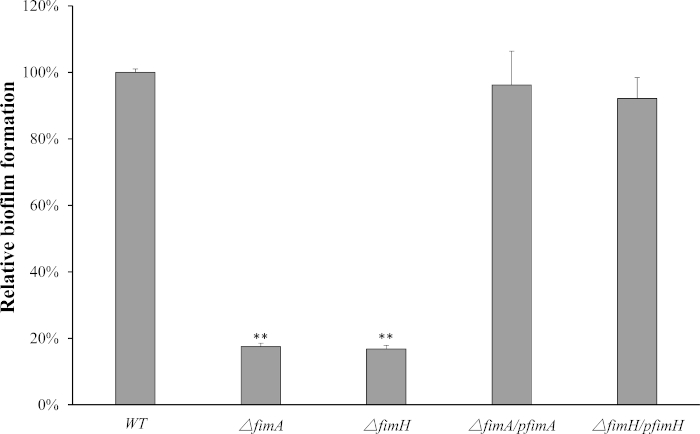
Figure 2: Type 1 fimbriae improved F18ab fimbriae+ STEC biofilm formation. Surface-adhered biofilm was quantified by measuring OD600 of ethanol-solubilized CV staining. Data is presented relative to the absorbance of the WT strain, which was normalized to 1.0. Mean and standard deviation of triplicate experiments are shown. Significant differences between the mutants and WT strain are indicated (* p < 0.05, ** p < 0.01). Please click here to view a larger version of this figure.
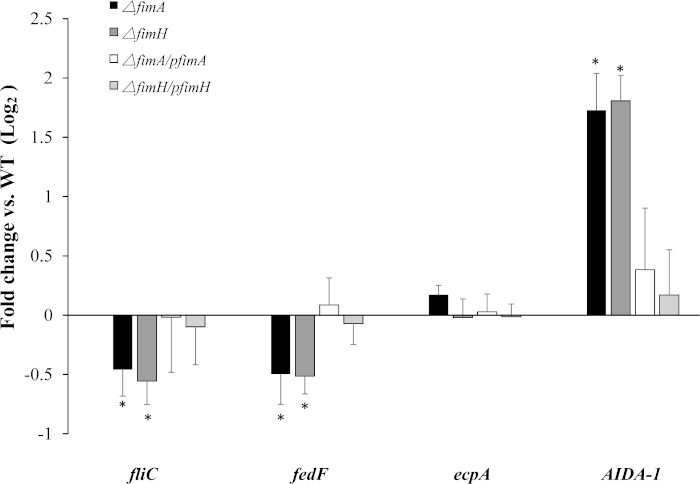
Figure 3: Deletion of fimA or fimH gene affects the expression of other adhesins in F18ab fimbriae+ STEC. gapA was used as the normalizing internal standard. Changes (n-fold) were calculated using WT F107/86 as the relative measure of comparison. Mean and standard deviation of triplicate experiments are shown. Significant differences between the mutants and WT strain are indicated (* p < 0.05). Please click here to view a larger version of this figure.
| Primer | Sequences (5’-3’) | Description | Reference | |
| gapA-RT-F | CGTTAAAGGCGCTAACTTCG | qPCR | 12 | |
| gapA-RT-R | ACGGTGGTCATCAGACCTTC | |||
| fedF-RT-F | CCGTTACTCTTGATTTCTTTGTTG | qPCR | 12 | |
| fedF-RT-R | GGCATTTGGGTAGTGTTTGTCTT | |||
| fliC-RT-F | ACTCAGAAAACCTGATGGTGAAACT | qPCR | 12 | |
| fliC-RT-R | CCCCACCTCTCCCTAACACA | |||
| ecpA-RT-F | CACTGAATGTGGGCGTTGAT | qPCR | In this study | |
| ecpA-RT-R | CTAAGGTTGCCGCCCAGTAC | |||
| AIDA-I-RT-F | CAGTCTACCGCACAAGCAAAAC | qPCR | 12 | |
| AIDA-I-RT-R | TCAATACACAAAACCCGATACCC | |||
Table 1: Primers used in this study.
Discussion
The methods provided here help to efficiently determine the function of fimbriae in bacterial colonization. Interestingly, in this study, deletion of fimA showed 15% less adhesion than fimH mutant, suggesting that tip adhesin may not be the only factor required for F18ab fimbriae+ STEC adhesion and that fimbrial rod subunit, FimA, works in bacterial attachment as well (Figure 1A). A recent study proposed that FimA modulated mechanical properties of the fimbrial shaft could exert a significant effect on E. coli adhesion under drag forces caused by flowing bodily fluids25. This was also shown for E. coli K12 type 1 fimbriae-mediated adhesion26, and the results support this hypothesis. Otherwise, we found that deleting fimA or fimH significantly decreased F18ab fimbriae+ STEC invasion, which demonstrated the invasive function of type 1 fimbriae (Figure 1B). Meanwhile, the 23% less invasion ability of fimA mutant than fimH mutant suggested the fimbrial rod mediated adhesion enhancing the chance for bacteria invading to host cells (Figure 1B). However, reports showed that type 1 fimbriae may not be associated with or even negatively regulate biofilm formation27,28. In the biofilm formation assay, we found that both of FimA and FimH subunits of type 1 fimbriae are important for F18ab fimbriae+ STEC biofilm formation (Figure 2).
Limitations of the methods include that the stable gene knock-out mutants are required for the functional analysis study; and for bacterial adhesion / invasion assay, cell lines used in the experiments should be correlated with pathogen as well as its natural infection sites. In order to understand the function of fimbriae or other virulence in the pathogen, the single gene knock-out mutant and its complemented strain were prepared before assays. λ-Red recombination system we used was a good choice as it is convenient to operate in both E. coli and Salmonella strains with the constructed plasmids, including pKD3, pKD4, pKD46, and pCP20, and the mutant is usually stable for further study. However, this system cannot meet the requirements for all Gram-positive and several Gram-negative bacteria strains. Along with the development of CRISPR-Cas system, we believe a universal gene knock-out system will be suitable for all species of bacteria in the future, which can be beneficial to perform the functional comparative experiments for single virulence factor. In addition, we used an epithelial cell line (IPEC-J2) derived from the jejunum of un-suckled 1-day-old piglets that does not express F18 receptors10, to study the role of type 1 fimbriae in F18ab fimbriae+ STEC adhesion and invasion, which not only mimicked intestinal environments but also ruled out the influence from F18 fimbriae. Therefore, for bacteria that have no correlated in vitro cell model, preparation of a stable primary cell line may be the major concern.
It is also important to note that the fimbrial gene knock-out in bacteria may result in co-regulation of other adhesins29. Thus, we performed the qPCR to determine the expression of several key adhesins in F18ab fimbriae+ STEC. The expression of fliC and fedF were downregulated by about 30% in the mutants, as compared with their expression in the WT strains. We previously demonstrated that it was flagella, but not F18 fimbriae, mediating F18ab fimbriae+ STEC adhesion and invasion to IPEC-J2 cells30, suggesting that reduced adhesion and invasion in the both mutants are due at least in part to the reduction in fliC expression. On the other hand, we observed up to three-fold increase of AIDA-I expression in the ΔfimA and ΔfimH mutants, bacterial adhesion and biofilm formation were still reduced, suggesting that type 1 fimbriae may affect much greater than autotransporter proteins in F18ab fimbriae+ STEC biofilm formation.
In summary, the methods described in this study provide a useful approach for determining the role of bacterial fimbriae or other virulence playing in the colonization. Future applications of these methods could advance by the development of universal bacterial gene knock-out system and ex-in vivo cell model for bacterial infection. Although the data here demonstrated the role of type 1 fimbriae, especially the rod subunit (FimA), in adhesion, invasion and biofilm formation of F18ab fimbriae+ STEC, a detailed molecular interaction between FimA / FimH and cellular receptor is required to confirm this using techniques such as pull-down and co-immunoprecipitation in the future.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (No. 31672579).
Materials
| 96-well microplate | Corning | 3599 | adhesion and invasion assay |
| 96-well microplate(Round bottom) | Corning | 3799 | biofilm formation |
| crystal violet | Sinopharm Chemical Reagent | 71012314 | Biofilm staining |
| dextrose | Sangon Biotech | A610219 | Culture broth |
| Ex Taq | TaKaRa | RR01A | PCR |
| F12 medium | Gibco | 11765062 | Cell culture |
| FeSO4 | Sangon Biotech | A501386 | Culture broth |
| K2HPO4 | Sinopharm Chemical Reagent | 20032116 | Culture broth |
| KH2PO4 | Sinopharm Chemical Reagent | 10017608 | Culture broth |
| L-Arabinose | Sangon Biotech | A610071 | λ-Red recombination |
| MgSO4 | Sinopharm Chemical Reagent | 20025117 | Culture broth |
| NaCl | Sinopharm Chemical Reagent | 10019308 | Culture broth |
| (NH4)2SO4 | Sinopharm Chemical Reagent | 10002917 | Culture broth |
| Micro spectrophotometer | Thermo Fisher | Nano Drop one | Nucleic acid concentration detection |
| New-born calf serum | Gibco | 16010159 | Cell culture |
| Peptone | Sangon Biotech | A505247 | Culture broth |
| PrimeScript RT reagent Kit with gDNA Eraser | TaKaRa | RR047 | qPCR |
| Real-Time PCR | Applied Biosystems | 7500 system | qPCR |
| RPMI1640 medium | Gibco | 11875500 | Cell culture |
| Spectrophotometer | Eppendorf | BioSpectrometer | Absorbance detection |
| Spectrophotometer (96-well microplate) | BioTek | Epoch | Absorbance detection |
| SYBR Premix Ex Taq II | TaKaRa | RR820 | qPCR |
| Tabletop centrifuge | Thermo Fisher | Micro 17(R) | Centrifugation |
| thiamine hydrochloride | Sangon Biotech | A500986 | Culture broth |
| Triton X-100 | Sangon Biotech | A110694 | adhesion and invasion assay |
| TRIzol | Invitrogen | 15596018 | RNA isolation |
| Tryptone | Oxoid | LP0042 | Culture broth |
| Yeast extract | Oxoid | LP0021 | Culture broth |
References
- Ofek, I., Beachey, E. H. Mannose binding and epithelial cell adherence of Escherichia coli. Infection and Immunity. 22 (1), 247-254 (1978).
- Khan, N. A., Kim, Y., Shin, S., Kim, K. S. FimH-mediated Escherichia coli K1 invasion of human brain microvascular endothelial cells. Cellular Microbiology. 9 (1), 169-178 (2007).
- Ashkar, A. A., et al. FimH adhesin of type 1 fimbriae is a potent inducer of innate antimicrobial responses which requires TLR4 and type 1 interferon signalling. PLoS Pathogens. 4 (12), (2008).
- Pusz, P., Bok, E., Mazurek, J., Stosik, M., Baldy-Chudzik, K. Type 1 fimbriae in commensal Escherichia coli derived from healthy humans. Acta Biochimica Polonica. 61 (2), 389-392 (2014).
- Gunther, N. W., Lockatell, V., Johnson, D. E., Mobley, H. L. In Vivo Dynamics of Type 1 fimbria regulation in uropathogenic Escherichia coli during experimental urinary tract infection. Infection and Immunity. 69 (5), 2838-2846 (2001).
- Justice, S. S., et al. Differentiation and developmental pathways of uropathogenic Escherichia coli in urinary tract pathogenesis. Proceedings of the National Academy of Sciences of the United States of America. 101 (5), 1333-1338 (2004).
- Imberechts, H., et al. Characterization of F18 fimbrial genes fedE and fedF involved in adhesion and length of enterotoxemic Escherichia coli strain 107/86. Microbial Pathogenesis. 21 (3), 183-192 (1996).
- Nagy, B., et al. Biological relationship between F18ab and F18ac fimbriae of enterotoxigenic and verotoxigenic Escherichia coli from weaned pigs with oedema disease or diarrhoea. Microbial Pathogenesis. 22 (1), 1-11 (1997).
- Ravi, M., et al. Contribution of AIDA-I to the pathogenicity of a porcine diarrheagenic Escherichia coli and to intestinal colonization through biofilm formation in pigs. Veterinary Microbiology. 120 (3-4), 308-319 (2007).
- Duan, Q., et al. The flagella of F18ab Escherichia coli is a virulence factor that contributes to infection in a IPEC-J2 cell model in vitro. Veterinary Microbiology. 160 (1-2), 132-140 (2012).
- Rendón, M. A., et al. Commensal and pathogenic Escherichia coli use a common pilus adherence factor for epithelial cell colonization. Proceedings of the National Academy of Sciences of the United States of America. 104 (25), 10637-10642 (2007).
- Duan, Q., Nandre, R., Zhou, M., Zhu, G. Type I fimbriae mediate in vitro adherence of porcine F18ac+ enterotoxigenic Escherichia coli (ETEC). Annals of Microbiology. 44 (1), (2017).
- Zeiner, S. A., Dwyer, B. E., Clegg, S. FimA, FimF, and FimH are necessary for assembly of type 1 fimbriae on Salmonella enterica Serovar Typhimurium. Infection and Immunity. 80 (9), 3289-3296 (2012).
- Datsenko, K. A., Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proceedings of the National Academy of Sciences of the United States of America. 97 (12), 6640-6645 (2000).
- Guo, Z. Study on FimA mediated F18ab+ Escherichi coli pathogenicity. Yangzhou University. , (2014).
- Kudva, I. T., Dean-Nystrom, E. A. Bovine recto-anal junction squamous epithelial (RSE) cell adhesion assay for studying Escherichia coli O157 adherence. Journal of Applied Microbiology. 111, 1283-1294 (2011).
- Berschneider, H. Development of normal cultured small intestinal epithelial cell lines which transport Na and Cl. Gastroenterology. 96, 41 (1989).
- Brosnahan, A. J., Brown, D. R. Porcine IPEC-J2 intestinal epithelial cells in microbiological investigations. Veterinary Microbiology. 156, 229-237 (2012).
- Koh, S. Y., et al. Porcine intestinal epithelial cell lines as a new in vitro model for studying adherence and pathogenesis of enterotoxigenic Escherichia coli. Veterinary Microbiology. 130, 191-197 (2008).
- Dogan, B., et al. Phylogroup and lpfA influence epithelial invasion by mastitis associated Escherichia coli. Veterinary Microbiology. 159, 163-170 (2012).
- Hossain, M. M., Tsuyumu, S. Flagella-mediated motility is required for biofilm formation by Erwinia carotovora subsp. carotovora. Journal of General Plant Pathology. 72 (1), 34-39 (2006).
- Wang, Y., Chen, H., Zhu, X. Observation on Pseudomonas aeruginosa biofilm with sliver staining method. Chinese Journal of Microecology. 12 (1), (2012).
- Ambalam, P., Kondepudi, K. K., Nilsson, I., Wadström, T., Ljungh, &. #. 1. 9. 7. ;. Bile stimulates cell surface hydrophobicity, congo red binding and biofilm formation of lactobacillus strains. FEMS Microbiology Letters. 333 (1), 10-19 (2012).
- Schmittgen, T. D., Livak, K. J. Analyzing real-time PCR data by the comparative C(T) method. Nature Protocols. 3 (6), (2008).
- Kisiela, D. I., et al. Evolutionary analysis points to divergent physiological roles of type 1 fimbriae in Salmonella and Escherichia coli. mBio. 4 (2), (2013).
- Forero, M., Yakovenko, O., Sokurenko, E. V., Thomas, W. E., Vogel, V. Uncoiling mechanics of Escherichia coli type I fimbriae are optimized for catch bonds. PLoS Biology. 4 (9), 298 (2006).
- Di Martino, P., Cafferini, N., Joly, B., Darfeuille-Michaud, A. Klebsiella pneumoniae type 3 pili facilitate adherence and biofilm formation on abiotic surfaces. Research in Microbiology. 154 (1), 9-16 (2003).
- Fazli, M., et al. The exopolysaccharide gene cluster Bcam1330-Bcam1341 is involved in Burkholderia cenocepacia biofilm formation, and its expression is regulated by c-di-GMP and Bcam1349. MicrobiologyOpen. 2 (1), 105-122 (2013).
- Zamani, H., Salehzadeh, A. Biofilm formation in uropathogenic Escherichia coli: association with adhesion factor genes. Turkish Journal of Medical Sciences. 48 (1), 162-167 (2018).
- Duan, Q., et al. Contribution of flagellin subunit FliC to piglet epithelial cells invasion by F18ab E. coli. Veterinary Microbiology. 166 (1-2), 220-224 (2013).

.