1. Isolate total RNA from plant tissues26,27
- Homogenize the plant tissues.
- Collect 100 mg of a fresh plant tissue (e.g., 4-week-old seedlings from Arabidopsis thaliana). Freeze the tissue and a pestle and mortar with liquid nitrogen, followed by grinding the tissue into powder.
- Add 1 mL of RNA isolation reagent (see Table of Materials) into the mortar. The reagent will be frozen immediately. Homogenize the tissue sample with the pestle when the frozen reagent melts.
- Transfer the homogenate to a 1.5-mL tube, centrifuge the sample at 12,000 x g for 5 min at 4 °C, and then transfer the cleared homogenate solution to another fresh 1.5-mL tube.
- Incubate the homogenized sample at room temperature for 5 min.
- Isolate total RNA.
- Add 0.2 mL of chloroform to the homogenate, cap the tube securely, shake the tube vigorously by hand for 15 s, and incubate the sample at room temperature for 5 min.
- Centrifuge the sample at 12,000 x g for 15 min at 4 °C and transfer the colorless upper aqueous phase to a fresh 1.5-mL tube. The sample separates into three phases following centrifugation.
- Add 0.5 mL of isopropyl alcohol to the aqueous phase, shake the tube by hand in a vigorous manner, and incubate the mixture at room temperature for 10 min.
- Centrifuge the mixture at 12,000 x g for 10 min at 4 °C and remove the supernatant.
- Wash the RNA pellet once with 1 mL of 75% ethanol by vortexing, followed by centrifugation at 7,500 x g for 5 min at 4 °C.
- Repeat step 1.2.5.
- Air dry the pellet for 5-10 minutes and redissolve the RNA in diethylpyrocarbonate (DEPC)-treated water by pipetting up and down, followed by measuring the total RNA concentration with a micro-spectrophotometer (see Table of Materials).
2. Synthesize complementary DNA (cDNA)28
- Synthesize the first strand of cDNA using a kit (see Table of Materials). Set up a 20-μL reaction system as shown in Table 1 and incubate the reaction tube in a PCR instrument for 50 min at 42 °C, followed by terminating the reaction at 85 °C for 5 min. Store the reaction product at -20 °C for future amplification of genes.
| Reagents | Volume |
| dNTP Mix, 2.5mM each | 4.0 μL |
| Primer Mix | 2.0 μL |
| RNA Template | 1.0 μg |
| Reverse Transcriptase Buffer, 5× | 4.0 μL |
| Reverse Transcriptase, 200 U/μL | 1.0 μL |
| RNase-Free H2O | up to 20.0 μL |
Table 1: Reverse transcription of total RNA into cDNA
3. Construct recombinant plasmids29
- Design PCR primers.
- Design the PCR primers using a software (see Table of Materials) based on the sequences of key enzyme genes obtained from the GenBank database and synthesize the primers by a company (see Table of Materials). In the 5’ end of the primer, add a restriction enzyme site (e.g., BamHI or EcoRI in this protocol).
NOTE: The primers used in this study are shown in Table 2.
- Design the PCR primers using a software (see Table of Materials) based on the sequences of key enzyme genes obtained from the GenBank database and synthesize the primers by a company (see Table of Materials). In the 5’ end of the primer, add a restriction enzyme site (e.g., BamHI or EcoRI in this protocol).
| Sequence, 5' → 3' | Purpose |
| AAGGATCCATGGCTCCAGGAACTTTGACT | Forward primer for PCR amplification of Atf3h gene from Arabidopsis thaliana. BamHI site is italicized and attached for cloning into pET32a(+). |
| AAGAATTCCTAAGCGAAGATTTGGTCGA | Reverse primer for PCR amplification of Atf3h gene from A. thaliana. EcoRI site is italicized and attached for cloning into pET32a(+). |
| AAGGATCCATGGAGGTCGAAAGAGTCCA | Forward primer for PCR amplification of Atfls1 gene from A. thaliana. BamHI site is italicized and attached for cloning into pET32a(+). |
| AAGAATTCTCAATCCAGAGGAAGTTTAT | Reverse primer for PCR amplification of Atfls1 gene from A. thaliana. EcoRI site is italicized and attached for cloning into pET32a(+). |
Table 2: Oligonucleotide primers used in the current study
- Clone the genes into a prokaryotic expression vector.
- Amplify the genes from the first strand of the synthesized cDNA using a high-fidelity DNA polymerase (see Table of Materials). Set up a 100-μL PCR reaction system as shown in Table 3 and run the following PCR cycle: 94 °C for 2 min for initial denaturation; then 35 cycles of 94 °C for 30 s for denaturation, 55 °C for 2 min for annealing, and 72 °C for 1 min for extension; followed by a final elongation at 72 °C for 10 min. Cool the reaction mixture to 12 °C.
NOTE: The extension time is variable and determined by the gene length with polymerization of about 1000 bases per min for most DNA polymerases. - Visualize the PCR products (most commonly 5 μL) on a 1% agarose gel and purify the specific DNA fragment from the remaining products using a DNA clean-up kit (see Table of Materials).
- Digest the purified DNA fragment and the vector (e.g., pET-32a(+)) with restriction enzymes (e.g., BamHI or EcoRI in this protocol). Set up a 50-μL reaction system in a 0.2 mL PCR tube as shown in Table 4 and incubate the mixture at 37 °C for 3 h. Separate the digested DNA on a 1% agarose gel.
- Recover the DNA band using a gel extraction kit (see Table of Materials). Further purify the DNA using a DNA clean-up kit (see Table of Materials), followed by measuring the concentration of DNA with a micro-spectrophotometer (see Table of Materials).
- Ligate the gene fragment into the linearized vector DNA using a T4 DNA ligase (see Table of Materials). Set up a ligation reaction in a 1.5-mL tube as shown in Table 5 and incubate the tube at room temperature for 2 – 3 h.
NOTE: The molar ratio of an insert to a vector is variable and ranged from 3:1 to 10:1. - Add 2.5 μL of the ligation mixture into 50 μL of chemically competent Escherichia coli cells (e.g., TOP10 or DH5α), mix gently, and keep the tube on ice for 30 min. Heat shock the cells at 42 °C for 90 s and immediately place the tube on ice for 2 min.
- Add 200 μL of liquid LB medium without antibiotics into the tube and incubate the tube in a 37 °C shaker at 220 rpm for 1 h. Spread 50 – 100 μL of the cells on an LB plate containing 100 μg/mL ampicillin and incubate at 37 °C overnight.
- Amplify the genes from the first strand of the synthesized cDNA using a high-fidelity DNA polymerase (see Table of Materials). Set up a 100-μL PCR reaction system as shown in Table 3 and run the following PCR cycle: 94 °C for 2 min for initial denaturation; then 35 cycles of 94 °C for 30 s for denaturation, 55 °C for 2 min for annealing, and 72 °C for 1 min for extension; followed by a final elongation at 72 °C for 10 min. Cool the reaction mixture to 12 °C.
| Reagents | Volume |
| Pfu Master Mix, 2× | 50.0 μL |
| Forward Primer, 10 μM | 4.0 μL |
| Reverse Primer, 10 μM | 4.0 μL |
| cDNA | 2.0 μL |
| H2O | 40.0 μL |
Table 3: Setting up of a PCR reaction system
| Reagents | Volume |
| DNA Fragment/Vector | 3.0 μg |
| BamHI | 1.0 μL |
| EcoRI | 1.0 μL |
| Cutsmart Buffer, 10× | 5.0 μL |
| H2O | up to 50.0 μL |
Table 4: Double digestion of a DNA fragment/vector
| Reagents | Volume |
| Insert | X μL (0.09 pmol) |
| Vector | Y μL (0.03 pmol) |
| Ligation Buffer, 10× | 1.0 μL |
| T4 DNA Ligase, 400 U/μL | 1.0 μL |
| H2O | up to 10.0 μL |
Table 5: Ligation of a gene fragment into a linearized vector
- Screen positive colonies.
- Inoculate a single colony from the LB plate into 200 μL of liquid LB medium containing 100 μg/mL ampicillin and incubate at 37 °C, 250 rpm for 2 – 3 h.
NOTE: In general, pick 4 – 8 colonies for screening positive colonies. - Set up a 10-μL colony PCR reaction similar to that in step 3.2.1.
NOTE: Use 1 μL of LB culture instead of 1 μL of cDNA template. - Visualize the PCR products on a 1% agarose gel. Inoculate the remaining culture with a positive result into 3 mL of liquid LB medium containing 100 μg/mL ampicillin and incubate in a 37 °C shaker at 250 rpm for 14 – 16 h.
- Isolate plasmid DNA from recombinant E. coli cultures using a plasmid miniprep kit (see Table of Materials).
- Identify the purified recombinant plasmids by a double restriction enzyme analysis (e.g., BamHI and EcoRI in this protocol). Set up a 10-μL reaction system similar to that in step 3.2.3, followed by incubation at 37 °C for 3 h. Visualize the specific band released from the recombinant plasmid on a 1% agarose gel.
- Inoculate a single colony from the LB plate into 200 μL of liquid LB medium containing 100 μg/mL ampicillin and incubate at 37 °C, 250 rpm for 2 – 3 h.
- Verify the sequences of positive recombinant plasmids.
- Send the plasmids to a company for sequencing. Analyze the results using a DNA sequence analysis software (see Table of Materials) by comparing the sequence obtained from the sequencing company with the reference sequence obtained from the GenBank database.
4. Express recombinant enzyme proteins30
- Transform the correct recombinant plasmid into competent E. coli BL21(DE3).
- Add 0.1 μL of the plasmid to 10 μL of competent E. coli BL21(DE3) in a 1.5-mL tube on ice and keep the tube on ice for 5 min.
- Heat shock the cells in a 42 °C waterbath for 90 s and place it on ice again for 2 min.
- Add 200 μL of LB liquid medium without antibiotics and incubate in a 37 °C shaker at 220 rpm for 5 min.
- Spread 50 μL of transformation on an LB agar plate containing 100 μg/mL ampicillin and incubate the plate overnight in a 37 °C incubator.
- Induce the expression of genes.
- Inoculate 3 – 5 colonies from the plate into a tube containing 3 mL of LB liquid medium with 100 μg/mL ampicillin and incubate at 250 rpm in a 37 °C shaker overnight.
- Transfer all of the overnight culture into 300 mL of LB liquid medium containing 100 μg/mL ampicillin and incubate at 250 rpm in a 37 °C shaker until the optical density of the culture at 600 nm is between 0.4 – 0.6.
- Add isopropyl β-D-thiogalactoside (IPTG) into the culture with a final concentration of 0.2 mM and induce the expression of the genes at 250 rpm, 20 – 22 °C for 3 h.
5. Purify the recombinant enzyme proteins31
- Harvest the bacteria by centrifugation of the culture at 4 °C, 12,000 x g for 10 min.
- Resuspend the pellet in 15 mL of Bacterial Lysis Buffer containing 0.1% Triton X-100, 1 mM EDTA, 10% glycerol, 150 mM NaCl, 0.5 mM DTT, 0.1 mM PMSF, 1 μg/mL aprotinin, 1 μg/mL leupeptin, and 1 μg/mL pepstatin in 50 mM Tris-Cl (pH 8.0).
- Sonicate the bacterial suspension to release the recombinant enzyme proteins, followed by centrifugation at 13,000 x g, 4 °C for 10 min.
- Harvest and aliquot the supernatant in 1.5-mL tubes at 1 mL/tube and store them at -70 °C for future use.
- Apply 500 μL of His-tag purification resin (see Table of Materials) to a reusable empty affinity column. Wash the resin with 5 bed volumes of deionized water to discard the ethanol in the stock solution. Balance the resin with 10 bed volumes of the binding buffer comprising Tris-Cl (20 mM, pH 7.9), imidazole (10 mM), and NaCl (0.5 M).
- Apply 4 mL of the supernatant from Step 5.4 to a slurry of the above resin and block two ends of the column with stoppers.
- Incubate the mixture at 4 °C on a rotator at low speed for 2 h.
- Wash the fusion protein bound resin with 15 bed volumes of the binding buffer at 4 °C at a flow rate of 1 mL/min before elution.
- Add 500 μL of elution buffer (containing 20 mM Tris-Cl (pH 7.9), 500 mM imidazole, 0.5 M NaCl) to the column and incubate the slurry at 4 °C on a rotator at a low speed for 10 min. Collect the eluent as purified protein samples.
- Repeat Step 5.5 four more times.
- Wash the resin sequentially with 10 bed volumes of deionized water and 3 bed volumes of 20% ethanol. Soak the resin in 20% ethanol. Block the column with stoppers and store it at 4 °C.
- Measure the concentration of the purified proteins by the Bradford protein assay. Determine the purity of the proteins on a 10% SDS-PAGE gel and visualize the bands by the Coomassie blue staining assay.
- Add glycerol to the purified protein solution to a final concentration of 10% to stabilize the enzyme activity. Aliquot and store it at -80 °C.
6. Produce a flavonol from a flavanone in an in vitro bienzyme synthetic system16
- Prepare buffers.
- Make 2x synthetic buffer without ferrous sulfate consisting of 200 mM Tris-HCl (pH 7.2), 16.4 mM α-ketoglutaric acid, 0.8% sodium ascorbate, and 20% glycerol. Dissolve 1.938 g of Tris base, 0.640 g of sodium ascorbate, 0.250 g of α-ketoglutaric acid, and 16 mL of glycerol to 64 mL of deionized water. Adjust pH to 7.2 by hydrochloric acid (HCl) and add deionized water up to 80 mL. Store the buffer at 4 °C for future use.
- Make a 100x stock solution of 2 mM ferrous sulfate. Dissolve 55.6 mg of ferrous sulfate heptahydrate in 50 mL of deionized water, stir, and add water up to 100 mL.
- Make a stock solution of 25 mM flavonoid. Dissolve a flavonoid in methanol thoroughly and stored at -20 ° C.
- Set up a synthetic system to produce a flavonol from a flavanone.
- Prepare the synthetic system as shown in Table 6.
- Incubate the reaction at 40 °C in an open 2.0-mL tube at 600 rpm (in a shaking heat block) for 40 min.
- Terminate the reaction by adding 10 μL of acetic acid and 100 μL of ethyl acetate.
- Two hours later, transfer the organic phases to 1.5-mL tubes for air drying in a hood at room temperature.
| Reagents | Volume |
| 2× Synthetic Buffer without ferrous sulfate | 50.0 μL |
| 25 mM flavonol | 2.0 μL |
| 2 mM ferrous sulfate | 0.5 μL |
| 1 mg/mL AtF3H | 2.5 μL |
| 1 mg/mL AtFLS1 | 2.5 μL |
| 25 mM flavanone | 2.0 μL |
| H2O | up to 100.0 μL |
Table 6: The synthetic system used in this protocol.
7. Analyze the reaction products
- Thin layer chromatography (TLC) analysis.
- Pool 5 tubes of the flavonoid samples from step 6.2.4 and take out 300 μL for air drying. Redissolve the flavonoid powder in 160 μL of methanol. Prepare authentic flavonoid samples with serial concentrations of 12.5, 25, 50, 100, and 200 ng/μL in methanol. Load 1 μL of the reaction samples and the authentic flavonoid samples onto polyamide 6 plates.
- Run the sample-loaded plates in a solvent system comprising chloroform/methanol/ethyl acetate/formic acid at a ratio of 5.0:1.5:1.0:0.5.
- Air dry the plates at room temperature. Spray the plates with 1% ethanolic solution of aluminum chloride (AlCl3), followed by air drying again at room temperature.
- Thirty minutes later, visualize the spots on the plates under a UV light at 254 nm and take images.
- Analyze the gray value of each spot on the images using an image processing software (e.g., ImageJ v1.51j8 in this protocol).
- Open the software ImageJ. Click File > Open to open the image to be analyzed.
- Click the left most Rectangular Selection Tool in the ImageJ User Interface. Outline the region of interest (ROI) in the image with the mouse and press
 to label the first ROI.
to label the first ROI. - Move the rectangular selection with the mouse right to the next ROI and press
 to label the second ROI.
to label the second ROI. - Repeat the previous step to label all other ROIs.
- Press
 to generate profile plots for all ROIs in a pop-up window.
to generate profile plots for all ROIs in a pop-up window.
NOTE: At this time, the Straight Line Selection Tool in the ImageJ User Interface will be automatically activated. - Use the Straight Line Selection Tool to draw base lines so as to define a closed area for each peak of interest.
- Activate the Wand Tool by clicking the corresponding icon in the ImageJ User Interface. Click inside the peak to display results for all peaks in a pop-up window.
- Make a TLC-based standard curve of the authentic flavonoid by plotting the gray values from step 7.1.5.7 against the corresponding flavonoid concentrations from step 7.1.1. Then, calculate the yield of the flavonoid of interest produced in this protocol according to the resulting formula.
- High performance liquid chromatography (HPLC) and liquid chromatography/mass spectrometry (LC/MS) analyses
- Process the samples from step 7.1.1 sequentially through 0.45 μm and 0.22 μm filters.
- Load the samples into a HPLC/LC/MS system (see Table of Materials) and separate the samples at 30 °C using a C18 (4.6 × 150 mm; i.d., 5 μm) column. Elute the column at 1.0 mL/min by a gradient of 10 – 85% (v/v) acetonitrile (ACN) in water (0 – 10 min, 10 – 25% ACN; 10 – 35 min, 25 – 50% ACN; 35 – 45 min, 50 – 85% ACN; 45 – 50 min, 85 – 10% ACN; 50 – 60 min, 10% ACN) and monitor the absorbance of the eluate from 200 to 800 nm. Perform the LC/MS analysis in a negative ion mode with a drying nitrogen flow of 10 L/min at 300 °C and a sheath gas flow of 7 L/min at 250 °C and collect data using a built-in software (see Table of Materials).
- Extract single wavelength chromatographs to calculate the peak areas of reaction samples and authentic flavonoid compounds using a software (see Table of Materials).
- Open the Qualitative Analysis program and click File > Open Data File. Select the file(s) to be analyzed in the Open Data File window and click Open to open the file(s).
- Right-click the mouse in the Chromatogram Results window and then the Extract Chromatograms in a pop-up menu.
- Open the Extract Chromatograms dialog box. In the Type list, click Other Chromatograms. In the Detector combo box, select DAD1. Then click OK to display the HPLC results in the Chromatogram Results window.
- Click the Manual Integration icon docked at the top of the Chromatogram Results window. Draw a base line for the peak required for manual integration analysis with the mouse.
- Click View > Integration Peak List to display the results.
- Make a HPLC-based standard curve of the authentic flavonoid by plotting the peak areas from step 7.2.3.5 against the corresponding flavonoid concentrations from step 7.2.1. Then, calculate the yield of the flavonoid of interest produced in this protocol according to the resulting formula.
- Analyze the MS data for the exact mass of flavonoid compounds using a software (see Table of Materials).
- Repeat steps 7.2.3.1 – 7.2.3.3.
- Click the Range Select icon on the Chromatogram Results toolbar.
- Select the peak of interest. Right-click the mouse in the selected range and click the Extract MS Spectrum in the pop-up menu to display the results in the MS Spectrum Results window.
F3H and FLS1 are two important key enzymes in the conversion of a flavanone into a flavonol in plants as shown in Figure 1. To develop an in vitro biosynthetic system for producing a flavonol from a flavanone, Atf3h (GenBank accession no.NM_114983.3) and Atfls1 (GenBank accession no. NM_120951.3) genes were cloned from the seedlings of 4-week-old A. thaliana into a prokaryotic expression vector pET-32a(+). The recombinant plasmids were transformed into E. coli BL21(DE3) and the fusion proteins were expressed after IPTG induction, followed by purification using Ni-IDA agarose resins. As shown in Figure 2, the purified fusion proteins showed a high purity of over 95% on a 10% SDS-PAGE gel, which were pure enough for the establishment of an in vitro bienzymatic cascade.
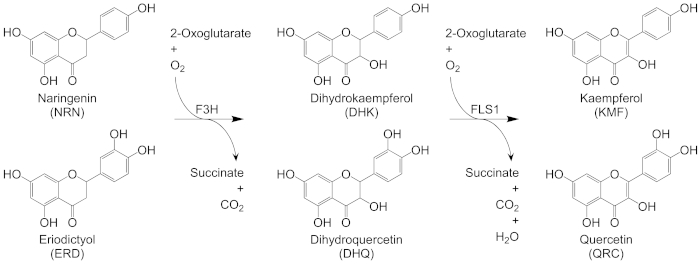
Figure 1: Schematic representation for the biosynthesis of a flavonol from a flavanonein vitro.F3H, flavanone 3-hydroxylase; FLS1, flavonol synthase 1. Please click here to view a larger version of this figure.
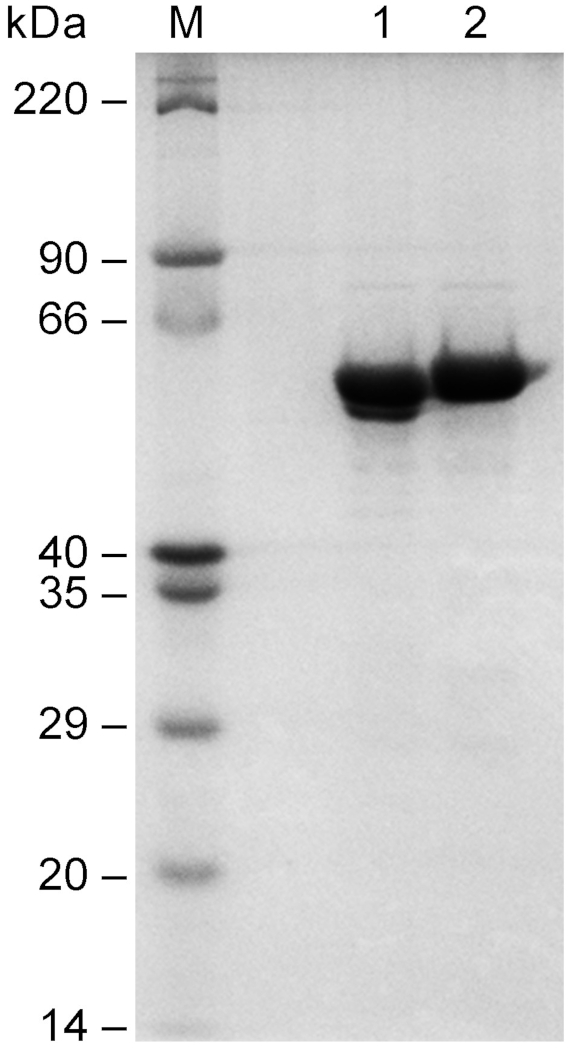
Figure 2: Purification of recombinant AtF3H and AtFLS1 proteins. The Atf3h and Atfls1 genes were cloned from 4-week-old seedlings of Arabidopsis thaliana into a prokaryotic expression vector pET-32a(+) and expressed in Escherichia coli BL21(DE3). The recombinant proteins were purified through an affinity chromatography column filled with Ni-IDA agarose resins. The purity was determined on a 10% SDS-PAGE gel. M, protein markers; 1, recombinant AtF3H protein; 2, recombinant AtFLS1 protein. Please click here to view a larger version of this figure.
To establish a bienzymatic cascade using the purified recombinant proteins, a synthetic system was prepared as shown in Table 6. To determine whether this system can be used for the conversion of a flavanone into a flavonol, NRN was added into the system, and the biosynthesis of KMF was detected by TLC and HPLC/LC/MS analyses. As shown in Figure 3A, there were two new spots emerged on a polyamide TLC plate. One spot showed a migration distance similar to that of dihydrokaempferol (DHK), and the other similar to that of KMF. Further analysis by HPLC and LC/MS demonstrated that the new chemicals showed a retention time of 11.91 min and 20.16 min, respectively (Figure 3B) and a quasi-molecular ion peak [M−H]− at m/z 287.0500 and 285.0500, respectively (Figure 3C), which were identical to those of DHK and KMF, respectively. The data indicate that KMF was produced from NRN in this system and the yield was as high as 34.94 mg/L.
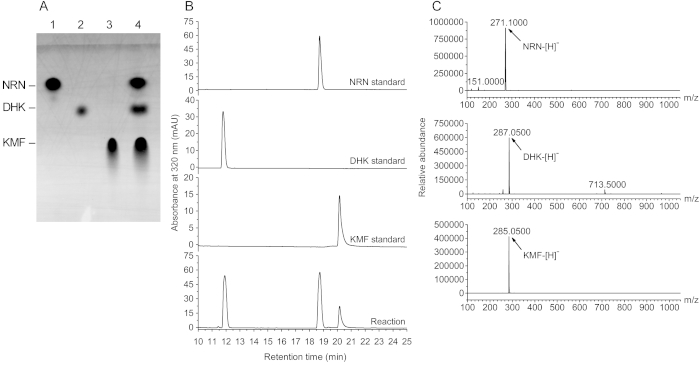
Figure 3: Synthesis of KMF from NRN in a bienzymatic cascade. (A) Analysis of the one-pot reaction products by polyamide TLC. 1, NRN standard; 2, DHK standard; 3, KMF standard; 4, reaction mixture. (B) HPLC analysis profiles of the reaction products. NRN, DHK, and KMF showed a retention time of 18.74 min, 11.91 min, and 20.16 min, respectively. (C) MS analysis profiles of the flavonoid compounds in the reaction mixtures. Please click here to view a larger version of this figure.
To further determine whether this in vitro system can be used for the conversion of other flavanones into their corresponding flavonols, eriodictyol (ERD) was added into the system to determine whether ERD can be converted into quercetin (QRC). As shown in Figure 4A, two new spots on a polyamide TLC plate displayed a migration distance similar to that of dihydroquercetin (DHQ) and QRC, respectively. HPLC and LC/MS analyses demonstrated that these new chemicals revealed a retention time of 10.03 min and 16.23 min, respectively (Figure 4B) and a quasi-molecular ion peak [M−H]− at m/z 303.1000 and 301.1000, respectively (Figure 4C), which exactly corresponded to those of DHQ and QRC, respectively. The data indicate that this system can convert ERD into QRC and the yield was 25.55 mg/L.
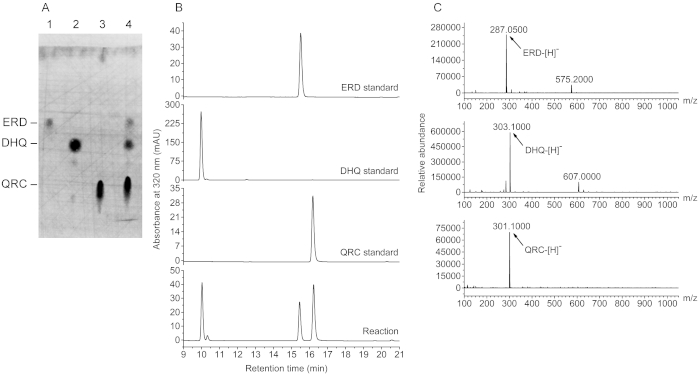
Figure 4: Production of QRC from ERD in a bienzyme synthetic system. (A) Analysis of the reaction products by polyamide TLC. 1, ERD standard; 2, DHQ standard; 3, QRC standard; 4, reaction mixture. (B) HPLC analysis profiles of the reaction products. ERD, DHQ, and QRC displayed a retention time of 15.45 min, 10.03 min, and 16.23 min, respectively. (C) MS analysis profiles of the compounds in the reaction mixtures. Please click here to view a larger version of this figure.
