We present a protocol for the preparation of synthetic cells by encapsulating a S30-T7 CFPS system based on BL21 E. coli inside lipid vesicles. A schematic description of the preparation process that includes an image of each stage is presented in Figure 2. The success of the synthetic cell preparation process is dependent on the appropriate performance of each stage and effected by different parameters. The protocol should be adjusted to accommodate the production of a specific protein.
Plasmids expressing the model protein super-folder Green Fluorescent Protein (sfGFP) and Renilla Luciferase under the T7 promoter were introduced into CFPS bulk reactions and synthetic cells, and protein production was evaluated using different methods, including western blot, flow cytometry, microscopy and spectroscopy (Figure 3). A verification of the sfGFP-His6 tagged protein (~27 kDa32) production by western blot analysis is presented in Figure 3A. A sample of 30 µL of synthetic cells diluted 6 folds was mixed with 10 µL of a common SDS-PAGE sample buffer (containing the detergents sodium dodecyl sulfate (SDS) and β-mercaptoethanol) and boiled for 10 min (95 ᵒC). We found that the combination of heat and detergents in the sample buffer is sufficient to disassemble the vesicles and to enable the running of proteins in the SDS-PAGE gel. Protein detection was performed using anti-His polyclonal primary antibody (diluted 1:12,000). As expected, while protein production is detected in samples containing sfGFP-encoding DNA templates ("+sfGFP DNA"), no protein is observed in negative control samples in which the DNA template was excluded ("-DNA"). Samples of purified sfGFP and purified sfGFP encapsulated within synthetic cells' membrane ('sfGFP (encapsulated)), are used as positive controls for the sfGFP CFPS bulk production and for its synthesis within synthetic cells, respectivley. This method can be applied to different protein types. According to Krinsky et al.19, the sfGFP production yield obtained under the detailed protocol is 380 µg/mL in CFPS solution and 5.3 µg/mL, when the CFPS solution is encapsulated inside lipid vesicles.
When the protein that is produced inside the synthetic cells is fluorescent, its production can be evaluated using microscopy and flow cytometry-based methods. We analyzed the synthetic cells using a fluorescent microscope with a filter for GFP fluorescence (Figure 3B). Since the CFPS components have some autofluorescent, the image acquisition parameters should be adapted according to an appropriate negative control sample (synthetic cells with no DNA template, for example).
In addition, we used flow cytometry to determine the mean fluorescence intensity of sfGFP-producing synthetic cells, and the percentage of active synthetic cells, which can produce proteins within them (Figure 3C I&II). 10,000 events were collected for each analyzed sample. The synthetic cell production, which is described in this protocol, usually yields an active population of 21-25% within a solution with an approximated concentration of 107 synthetic cells/mL. The slight fluorescence intensity presented by “-DNA” samples in Figure 3C II is due to the autofluorescence of different components in the CFPS reaction such as the S30 lysate.
Representative size analysis based on the GFP signal (in diameter) of synthetic cells prepared by the method described above shows a mean value of 2.4±0.5 µm (Figure 3D II). As the formation of water in oil emulsion in this method is an outcome of applying mechanical forces, the size distribution of the particles might be affected by different factors, such as the method and speed of pipetting the emulsion up-and-down, the model of the vortex mixer machine, etc.
To test the versatility of synthetic cells’ protein production, the expression of the reporter protein Renilla luciferase inside synthetic cells was analyzed (Figure 3E). The assay quantified Renilla luciferase activity by measuring the luminescence generated from the enzymatic reaction of luciferase and its substrate, h-coelenterazine. To induce the enzymatic reaction, a final concentration of 1 µM of h-coelenterazine was added to the synthetic cells (25 µL sample volume) just before the measurement. “-DNA” sample, not containing the luciferase-encoding DNA template was used as a negative control for testing luminescence due to non-enzymatic oxidation of the substrate. Luminescence was measured using a plate reader.
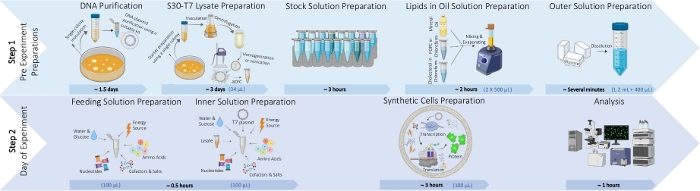
Figure 1: An illustration of the process for typical synthetic cells preparation protocol. The process is divided into two steps. Step 1: Pre-experiment preparations including DNA plasmid purification, S30-T7 lysate preparation, stock solutions preparation required for the inner and feeding solutions, lipids-in-oil solution preparation and outer solution preparation. Step 2: Synthetic cell formation that includes feeding and inner solutions preparation, synthetic cells preparation and analysis. An example of the required volume of each ingredient for preparing 100 µL of synthetic cells solution is presented in blue in brackets. Please click here to view a larger version of this figure.
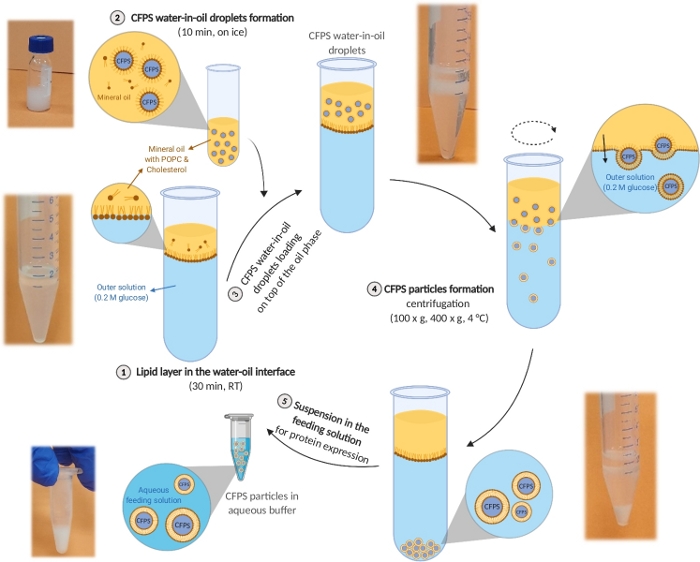
Figure 2: Schematic illustration of the synthetic cell preparation protocol. An image of a well-performed process illustrates each stage of the protocol. This figure has been modified from Krinsky et al.19. Please click here to view a larger version of this figure.
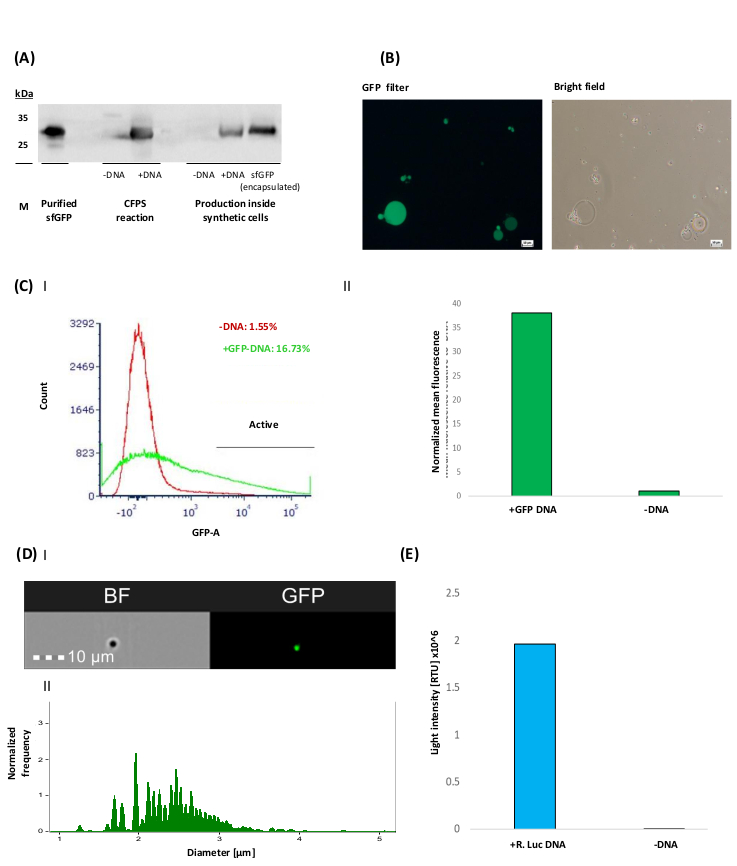
Figure 3: Representative results of the production of sfGFP and Renilla luciferase inside synthetic cells. (A) A western blot analysis of sfGFP-His6 production in both CFPS bulk reactions and inside synthetic cells. Protein detection was carried out using anti-His polyclonal primary antibody (diluted 1:12,000). Purified sfGFP-His6 was used as a positive control (7.8 µg). sfGFP (encapsulated) is positive control of purified sfGFP encapsulated in synthetic cells. Samples without DNA templates were used as a negative control for the production analysis ("-DNA"). (B) Representative images of sfGFP producing synthetic cells taken using a fluorescent microscope with a bright-field and a GFP filter. Scale bars = 50 µm. (C) I&II Flow cytometry analysis of sfGFP producing synthetic cells activity (Data collected using digital, 4-laser analyzer). Samples were measured after 3 h of incubation. 10,000 events were collected for each analyzed sample. FSC (500 V) and SSC (300 V) filters were used to define the total synthetic cell population. Then, a FITC (600 V) filter was used to detect GFP fluorescence.. (I) Calculation of the active synthetic cell population [%] was based on GFP fluorescence intensity threshold, defined by the “-DNA” sample (red histogram), allowing an error of ~1%. (II) Mean fluorescence intensity of sfGFP producing synthetic cells. This value was calculated from the active synthetic cell population and normalized to the “-DNA” sample by dividing the mean fluorescence of the active synthetic cells by the mean of the synthetic cells without sfGFP-DNA. (D) I&II Synthetic cell size analysis. The emission spectrum was detected by 505-560 nm and 642-745 nm for GFP and bright field signals, respectively. (I) A representative image of the analyzed synthetic cells. (II) Synthetic cells’ size distribution. Analysis was performed and the diameter distributions of the active synthetic cells was calculated based on the GFP signal. (E) Production of active Renilla luciferase inside synthetic cells. Luciferase activity was quantified with luminescence measurements after the addition of 1 µM h-coelenterazine using a plate reader and presented as light intensity [RLU] x 106. Please click here to view a larger version of this figure.
| Table 1: Buffer and stock solutions preparation. | |
| Comments | |
| Luria Bertani (LB) agar (1.5%) plate: | Prepare and sterilize. Add Ampicillin at a final concentration of 50 µg/mL only after cooling to room temperature. |
| 10 g/L Bacto-tryptone | |
| 10 g/L Sodium chloride (NaCl) | |
| 5 g/L Bacto-Yeast extract | |
| 15 g/L Agar agar purified | |
| 50 µg/mL Ampicillin | |
| LB media (20 mL): | Minimal media. Prepare and sterilize in advance. Just prior to inoculating the bacteria, add Ampicillin at a final concentration of 50 µg/mL. |
| 10 g/L Bacto-tryptone | |
| 10 g/L Sodium chloride (NaCl) | |
| 5 g/L Bacto-Yeast extract | |
| 50 µg/mL Ampicillin | |
| Terrific Broth (TB) media (1 L): | Rich media. Prepare and sterilize in advance. Just prior to inoculating the bacteria, add Ampicillin at a final concentration of 50 µg/mL. |
| 12 g/L Bacto-tryptone | |
| 24 g/L Bacto-Yeast extract | |
| 4% (v/v) Glycerol anhydrous | |
| 2.32 g/L K2HPO4 | |
| 12.54 g/L KH2PO4 | |
| 50 µg/mL Ampicillin | |
| S30 lysate buffer (1.5 L): | Prepare and sterilize in advance (*excluding DTT and 2-mercaptoethanol). Store at -4 °C. |
| 10 mM Tris-acetate at pH = 7.4 | Trisma-base. Prepare and adjust pH to 7.4 using Acetic acid. |
| 14 mM magnesium acetate | |
| 60 mM potassium acetate | |
| *1 mM DTT | Add just prior to use |
| *0.5 mL/L 2-mercaptoethanol | Add just prior to use |
| 1 M HEPES-KOH (pH = 8): | Dissolve HEPES to a final concentration of 1 M. Adjust to pH 8 using KOH solution. |
| HEPES | |
| Potassium hydroxide (KOH) | |
| Table 2: Inner solution composition. | ||||||||||
| Final requested Inner solution volume | ||||||||||
| Number | Reagent | Stock conc. | Final conc. | 25 µL | 50 µL | 100 µL | 200 µL | 300 µL | 400 µL | |
| 1 | HEPES KOH pH=8 | 1 M | 55 mM | 1.375 | 2.75 | 5.5 | 11 | 16.5 | 22 | Pre-Inner solution |
| 2 | Magnesium acetate | 1 M | 14 mM | 0.35 | 0.7 | 1.4 | 2.8 | 4.2 | 5.6 | |
| 3 | Potassium acetate | 1 M | 50 mM | 1.25 | 2.5 | 5 | 10 | 15 | 20 | |
| 4 | Ammonium acetate | 5.2 M | 155 mM | 0.75 | 1.5 | 3 | 6 | 9 | 12 | |
| 5 | PEG 6000 | 50% | 3% | 1.5 | 3 | 6 | 12 | 18 | 24 | |
| 6 | 3-PGA | 0.5 M | 40 mM | 2 | 4 | 8 | 16 | 24 | 32 | |
| 7 | Amino acids – mixture I | 50 M | 2.5 mM | 1.25 | 2.5 | 5 | 10 | 15 | 20 | |
| 8 | Amino acids – mixture II | 50 M | 2.5 mM | 1.25 | 2.5 | 5 | 10 | 15 | 20 | |
| 9 | ATP | 100 mM | 1.2 mM | 0.3 | 0.6 | 1.2 | 2.4 | 3.6 | 4.8 | |
| 10 | GTP | 50 mM | 1 mM | 0.5 | 1 | 2 | 4 | 6 | 8 | |
| 11 | UTP | 100 mM | 0.8 mM | 0.2 | 0.4 | 0.8 | 1.6 | 2.4 | 3.2 | |
| 12 | IPTG | 100 mM | 1 mM | 0.25 | 0.5 | 1 | 2 | 3 | 4 | |
| 13 | Sucrose | 2 M | 200 mM | 2.5 | 5 | 10 | 20 | 30 | 40 | |
| 14 ** | H2O UPW | ** | ** | ** | ** | ** | ** | |||
| 15 | S30-T7 Lysate | 34% | 8.5 | 17 | 34 | 68 | 102 | 136 | ||
| 16 * | DNA plasmid | 10 µg/mL | * | * | * | * | * | * | ||
| Table 3: Feeding solution composition. | |||||||||
| Final requested feeding solution volume | |||||||||
| Number | Reagent | stock conc. | final conc. | 25 µL | 50 µL | 100 µL | 200 µL | 300 µL | 400 µL |
| 1 | HEPES KOH pH=8 | 1 M | 83.3 mM | 2.08 | 4.17 | 8.33 | 16.67 | 25 | 33.33 |
| 2 | Magnesium acetate | 1 M | 21.2 mM | 0.53 | 1.06 | 2.12 | 4.24 | 6.36 | 8.48 |
| 3 | Potassium acetate | 1 M | 75.5 mM | 1.89 | 3.78 | 7.55 | 15.1 | 22.66 | 30.2 |
| 4 | Ammonium acetate | 5.2 M | 236.4 mM | 1.14 | 2.27 | 4.55 | 9.09 | 13.64 | 18.18 |
| 5 | PEG – 6000 | 50% | 4.54% | 2.27 | 4.54 | 9.08 | 18.16 | 27.24 | 36.32 |
| 6 | 3-PGA | 0.5 M | 60.1 mM | 3 | 6.01 | 12.01 | 24.02 | 36.04 | 48.04 |
| 7 | Amino acids – mixture I | 50 mM | 3.8 mM | 1.89 | 3.78 | 7.56 | 15.12 | 22.68 | 30.24 |
| 8 | Amino acids – mixture II | 50 mM | 3.8 mM | 1.89 | 3.78 | 7.56 | 15.12 | 22.68 | 30.24 |
| 9 | ATP | 100 mM | 1.8 mM | 0.45 | 0.91 | 1.81 | 3.62 | 5.43 | 7.24 |
| 10 | GTP | 50 mM | 1.5 mM | 0.76 | 1.51 | 3.02 | 6.04 | 9.06 | 12.08 |
| 11 | UTP | 100 mM | 1.2 mM | 0.3 | 0.61 | 1.21 | 2.42 | 3.63 | 4.84 |
| 12 | IPTG | 100 mM | 1.5 mM | 0.38 | 0.76 | 1.51 | 3.02 | 4.53 | 6.04 |
| 13 | Glucose | 2 M | 200 mM | 2.5 | 5 | 10 | 20 | 30 | 40 |
| 14 | H2O UPW | 5.92 | 11.83 | 23.7 | 47.38 | 71.06 | 94.76 | ||
| Table 4: Required solutions for synthetic cells preparation. | |
| Solution & materials required | Comments |
| Lipids in oil | Store at room temperature until use. Prepared according to section 2 |
| Outer solution | Prepared according to section 3.1 |
| Inner solution | Prepared according to section 3.2. |
| Prepare tests + controls samples according to section 4.1. | |
| Keep on crushed ice until use. | |
| Feeding solution | Prepared according to section 3.3. |
| Keep on crushed ice until use. | |
