Mosquitoes containing mutations in the genes AaeZIP11 (putative iron transporter21) and myo-fem (a female-biased myosin gene related to flight muscles13) were obtained using CRISPR/Cas9 technology, genotyped using HRMA, and sequence-verified (Figure 5). Figure 5A and Figure 5C show the normalized fluorescence intensity from the HRM curves from AaeZIP11 and myo-fem mutant samples, respectively, along with wild-type controls. Figure 4B and Figure 4D show the magnification of the difference curves (from the AaeZIP11 and myo-fem mutant samples, respectively) between the melt profiles of the different clusters assigned by the software after subtracting each curve from the wild-type reference. Heterozygous and homozygous mutant AaeZIP11 individuals were placed in different clusters and are easily distinguished from the wild-type controls (Figure 5B). Heterozygous mutant myo-fem individuals are also distinct from the controls (Figure 5D). Note that two clusters within the wild-type controls were present in both cases, most likely due to the presence of SNPs in the target region (Figure 5), highlighting the need to use multiple control samples to avoid categorizing controls as mutants when SNPs cannot be avoided.
Figure 4A shows the sequence analysis of the AaeZIP11 mutant. The electropherogram from AaeZIP11 heterozygous mutants indicates the nucleotide position where the indel occurred. This is represented by a shift from single to double peaks as polymorphic positions will show both nucleotides concomitantly (Figure 4A). The number of base pairs deleted or inserted was calculated by counting the single peaks at the end of the run (Figure 4B) as one of the DNA strands will be shorter or longer than the other by the number of base pairs deleted or inserted, respectively. It is recommended to use sequence-verified gDNA from the mutants as a reference for the identification of the heterozygotes to help with HRMA analyses for subsequent generations. Manual assignment for the curves may be needed when similar curves are automatically assigned to different clusters. This can be done by comparing the temperature-shifted curves and difference curves. Manual adjustment of the software might be needed to properly assign samples to clusters. HRMA results from AaeZIP11 and a mutant called Aeflightin show that individual sample analyses were needed to successfully categorize heterozygotes, homozygotes, and trans-heterozygotes. Initially, the automatic cluster assignment from the software could not make a proper distinction between the groups (Figure 6A, Figure 6C, Figure 6E, and Figure 6G). Each sample was then analyzed individually and assigned to the correct groups based on similarity to reference samples of heterozygotes, homozygotes, and trans-heterozygotes (previously verified by sequencing) (Figure 6B, Figure 6D, Figure 6F, and Figure 6H).

Figure 1: SNP identification. Schematic representation of multiple sequence alignment of AaeZIP11 fragment from wild-type. In red are the SNPs, and in green are the fragments free of SNPs; this SNP-free region is suggested for sgRNA and primer design. Abbreviations: SNP = single nucleotide polymorphism; sgRNA = single guide RNA; LVP = Liverpool strain. Please click here to view a larger version of this figure.

Figure 2: Experimental procedure for obtaining genomic DNA from mosquito legs for HRMA. (A) Materials for obtaining genomic DNA including pipettes, tips, PCR plate, optical seal, reservoir, the dilution buffer, and DNA release solution. (B) PCR plate preparation containing DNA release reagent and dilution buffer. (C) Mosquito anesthesia with CO2. (D) Wiping the tweezers with 70% ethanol to prevent contamination between samples. (E) Experimental setup including tweezers, rack for mosquito vials, Petri dish on top of an ice container with anesthetized mosquitoes, and the previously prepared PCR plate. (F) Removal of the mosquito leg. (G) Zoom view of the mosquito leg being submerged in the DNA release reagent solution. (H) Single mosquito placed in the vial. (I) Sealing the PCR plate containing the mosquito legs. Abbreviation: HRMA = high-resolution melt analysis. Please click here to view a larger version of this figure.
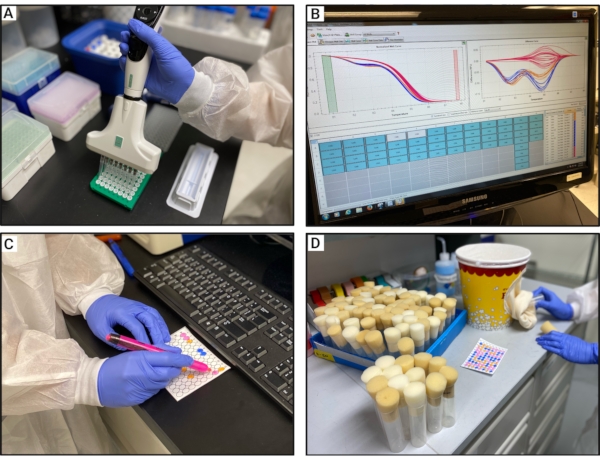
Figure 3: Experimental procedure for HRM analyses. (A) Transferring the released gDNA to a 96-well plate containing the PCR mix. (B) Visual inspection of the difference curves. (C) Marking each sample on the 96-well template with the same color as its respective difference curve color. (D) Backcrossing the individuals from the same cluster. Abbreviations: HRM = high-resolution melt; gDNA = genomic DNA. Please click here to view a larger version of this figure.
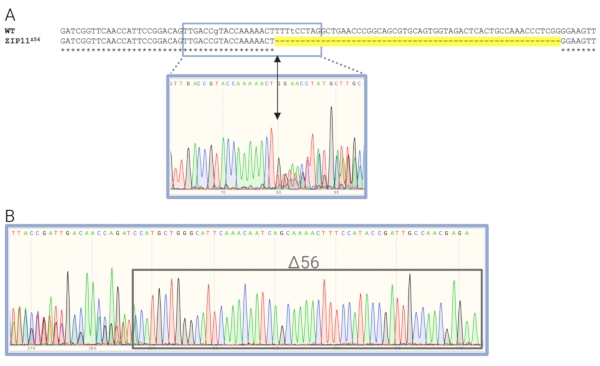
Figure 4: Sequence analysis of an AaeZIP11 mutant. (A) Nucleotide alignment of the AaeZIP11Δ56 mutant. Dashes highlighted in yellow are the deleted bases. In the box, the electropherogram transitions from single peaks to double peaks, depicting the position where the deletion occurred (arrow). (B) Electropherogram of the end of the sequencing run and the transition from double peaks to single peaks. Note that the number of single peaks represents the number of bases deleted (gray rectangle). Primer sequences are provided in the Supplemental Table S1. Please click here to view a larger version of this figure.
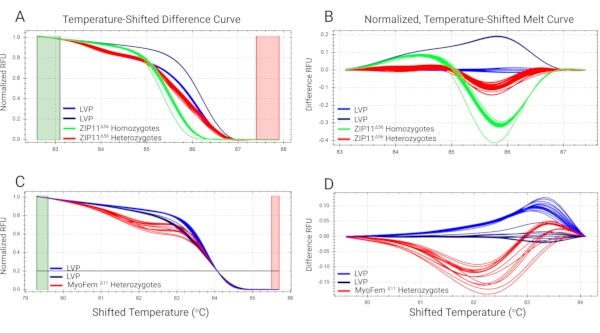
Figure 5: HRMA of DNA extracted from mosquito legs. DNA was extracted from a single leg from AaeZIP11 (A and B) and myo-fem (C and D) knockout Ae. aegypti mosquitoes and analyzed by HRMA. A and C denote the normalized fluorescence signals of the samples to relative values of 1.0 to 0. B and D denote the magnification of the curve differences by subtracting each curve from the wild-type reference (Liverpool strain). Abbreviations: HRMA = high-resolution melt analysis; LVP = Liverpool strain; RFU = relative fluorescence units. Primer sequences are provided in the Supplemental Table S1. Please click here to view a larger version of this figure.
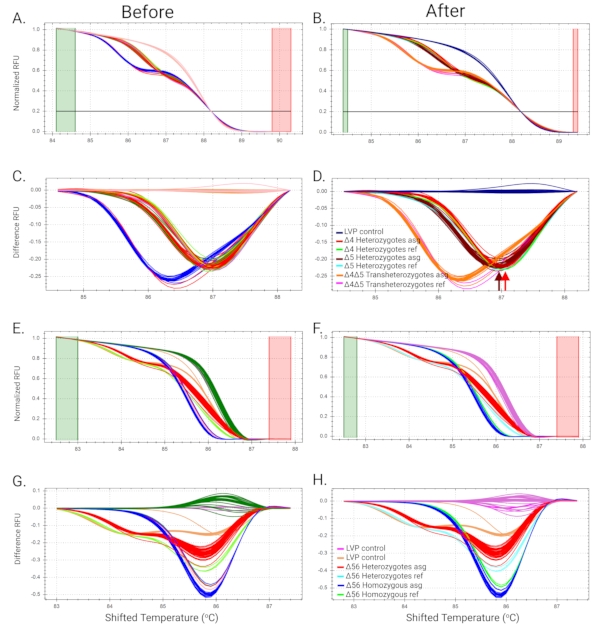
Figure 6: Examples of manual group assignments. (A–D) HRMA for Aeflightin mutants. (A and C) Melt curves (normalized linear scale curves and difference curves, respectively) are automatically grouped by the Precision Melt Analysis Software. (B) Normalization of differential curves altered manually. (D) After altering the normalization of the differential curves, each sample was assigned individually by the peak temperatures in the difference curves for corresponding positive controls (previously sequenced samples identified by Δ4 and Δ5 heterozygotes and Δ4Δ5 trans-heterozygotes), enabling the clear identification of the 3 groups. Red and brown arrows are added to highlight the peaks at different temperatures. (E–H) HRMA for AaeZIP11 mutants. (E and G) Melt curves are automatically assigned by the software. (F and H) Mutants in the second heterozygous self-cross were appropriately assigned to the correct groups by the similarity of the normalized and difference curves to previously determined references (color-coded in curves). Heterozygotes asg, Homozygotes asg, and Trans-heterozygotes asg: assigned melt curves by Precision Melt Analysis Software. Abbreviations: HRMA = high-resolution melt analysis; LVP = Liverpool strain; RFU = relative fluorescence units. Primer sequences are provided in Supplemental Table S1. Please click here to view a larger version of this figure.
Supplemental Material: Detailed protocol for setting up HRMA in the CFX96 Real-Time System (e.g., Bio-rad). Please click here to download this File.
Supplemental Figure S1: Step-by-step instructions for setting up the cycling protocol on Bio-rad CFX Manager. See Supplemental Material 2.1-2.3. Please click here to download this File.
Supplemental Figure S2: Step-by-step instructions for a plate setup on Bio-rad CFX Manager. See Supplemental Material 3.1-3.4. Please click here to download this File.
Supplemental Figure S3: Step-by-step instructions for a plate setup on Bio-rad CFX Manager. See Supplemental Material 3.5-3.6. Please click here to download this File.
Supplemental Figure S4: Step-by-step instructions for the run setup on Bio-rad CFX Manager. See Supplemental Material 4.1. Please click here to download this File.
Supplemental Figure S5: Step-by-step instructions for HRMA analysis on Bio-rad CFX Manager. See Supplemental Material 5.1-5.3. Abbreviation: HRMA = high-resolution melt analysis. Please click here to download this File.
Supplemental Table S1: List of primers. Please click here to download this Table.
