To perform the PK APAP tests in the 2-OC MPS, the first step is to manufacture the human intestine and liver equivalents (organoids). They are integrated into the 2-OC microfluidic device (Figure 1A) 24 h before starting of the PK APAP assay. The next day, the medium is changed, and the model is exposed to APAP. Figure 1 illustrates the intestine and liver equivalents placed inside the 2-OC device (Figure 1B) and the APAP PK experiment time course (Figure 1C). We performed a MTT assay, TEER measurements, HCA, real-time PCR, western blotting, histology, and confocal fluorescence microscopy in 2D culture and 3D organoids to check the tissue’s viability and detect possible APAP toxic effects. In the confocal fluorescence microscopy images, the intestine equivalent samples stained with DAPI and Phalloidin (for nuclei and actin respectively) were presented as a contiguous barrier for the non-treated (Figure 2A) and the 12 µM APAP treated samples (Figure 2B). As seen in Figure 2C, the MTT assay showed relative cell viability levels above 70%, indicating the absence of relevant cytotoxic effects in response to APAP exposure at 12 µM concentration33,34,35,36,37. The positive control (100 mM) induced significant cell death (survival below 5%). The Caco-2/HT-29 viability and proper differentiation as well as the intestine equivalent barrier integrity were verified by TEER evolution during the differentiation period (Figure 2D). APAP did not cause any alteration in the TEER values as shown in Figure 2E. The expression of the active sodium-coupled glucose transporters SLC5A1, multidrug resistance transporter MDR1 and sodium-potassium ATPase were analyzed, to verify the APAP treatment impact over cell barriers formation and basal functionality. As demonstrated in Figure 2F–H, both non-treated and APAP treated intestine equivalents have shown similar expression of SLC5A1 and NaKATPase. The oral administration of 12 µM APAP induced marked an increase in the MDR1 mRNA levels in intestines equivalents after 24 h (Figure 2H). We also performed HCA of cell phenotypic changes by a fluorophore dye mixture for nuclei and mitochondrial mass content. Positive controls were 100 mM APAP and 1% NaOH.
Additionally, we analyzed whether 12 µM APAP could induce cytotoxicity to the 2D Caco-2/HT-29 co-culture. The intestinal cells images acquired with fluorescence microscopy shown in Figure 2I corroborates the MTT data, which have demonstrated that 12 µM APAP did not cause significant cytotoxicity in the Caco-2/HT-29 intestinal equivalents.
The assessment of hepatic spheroids basal viability and the cytotoxicity in response to 2 µM APAP were done by MTT assay and morphological analyses by confocal fluorescence microscopy, H&E histology, and HCA assays. As shown in Figure 3A, the MTT assay was unable to identify any relevant cytotoxicity in response to the 2 μm APA treatment in samples taken from the Liver 2-OC assembly under both static and dynamic conditions. The cell viability decreased but remained over 80% for both 12 h and 24 h time-points33,34,35,36,37. The positive control treatments (100 mM APAP and 1% NaOH) induced significant tissue damages (viability below 10%). Microscopic confocal images indicate the absence of a necrotic center in the liver spheroids in both basal or APAP treatment conditions, and no evidence of significant death rates (Figure 3B-C). However, when we analyzed multiple cellular phenotypic changes following the vehicle or 2 µM APAP administration in the 2D HepaRG/HHSteC coculture through HCA assay, using 100 mM APAP (C+) as a positive control, contradictorily to the results of the MTT assay, the hepatic cells demonstrated early cytotoxic responses to 2 µM APAP treatment (Figure 3D). After 24 h, there was a decrease in the number of cells, in the nuclear area and an increase in mitochondrial mass. Additionally, a fluorophore dye cocktail containing Hoechst 33342 and MitoTracker Deep Red was used to stain the 3D hepatic spheroids (Figure 3J). Fiji software was used to evaluate 3D spherical architecture homogeneity among several spheroids (Figure 3E–I). The graphic shown in Figure 3E shows the similarity among liver spheroids total area. The aspect ratio (Figure 3F) around 1 means an absence of bias during the confection process of the spheroids. The evaluation also indicated that the majority of the spheroids were roughly rounded (Figure 3G). The evaluation of the morphology perimeter and cell distribution was done by circularity (Figure 3I) and by solidity calculation (Figure 3H), respectively. We concluded that the methodology to confectioning the liver spheroids had generated organoids with a smooth perimeter, compatible with spherical growth, no biases, or necrosis during the process.
As demonstrated in Figure 4A-B, the liver spheroids showed a relative basal high level of albumin and GST mRNA expression respectively, indicating proper basal functionality. Nevertheless, the 2 µM APAP treatment for 24 h induced a decrease in the albumin and the GST mRNA expression levels, suggesting impairment of liver spheroids functionally at 24 h time point of APAP treatment.
The detection of the CYP3A4 and UGT1A1 mRNA expression levels demonstrates the liver equivalents metabolic capacity. The CYP3A4 mRNA basal level (Figure 4C) was consistent with previous reports6. The APAP treatment induced a trend for a decrease in both CYP3A4 mRNA and UGT1A1 expression in response to APAP treatment (Figure 4C-D) once again corroborating the hypothesis of impairment of liver spheroids functionally at 24 h time point of APAP treatment.
Additionally, experiments of western blotting and in vitro enzymatic activity were performed in order to analyze the albumin protein expression, as well as, the CYP 3A4 activity in liver equivalents at basal and at APAP treatment conditions. We found that 2 µM APAP treatment performed at the Liver 2-OC MPS, induced a reduction in the liver equivalent total albumin expression at 12 h and 24 h time-points at both static (Figure 4E) and dynamic (Figure 4F) conditions. On the other hand, liver equivalents samples from dynamic conditions demonstrated a trend to present higher levels of protein expression of albumin when compared to the static conditions (Figure 4G). CYP 3A4 in vitro assay performed at liver equivalents from Liver 2-OC MPS shown that 2 µM APAP treatment for 12 h or 24 h was capable to induce a robust and significant impairment of CYP 3A4 activity at both static and dynamic conditions (Figure 4H-I). More interesting, the presence of media flow (dynamic) has induced a significant improvement of liver equivalents CYP 3A4 activity levels when compared to conditions in which the liver equivalents were kept in absence of circulating medium (static conditions) (Figure 4J).
To find the most sensitive analytical condition regarding HPLC analysis, several parameters were investigated including the composition of the mobile phase, the type, and concentration of additives. It was found that acetonitrile gave better chromatogram resolution and appropriate retention time than methanol. Fast and reproducible separation of APAP was obtained using a C18 reversed-phase column. The APAP retention time (Rt) value was 9.27 ± 0.19 minutes. Selectivity for APAP is indicated by the shape and symmetrical resolution of the peak, as well as by the lack of interfering peaks from the DMEM and Williams cell culture media.
APAP standard concentrations in DMEM S and Williams E S cell culture media diluted with ammonium acetate buffer (1:1, v/v) ranging from 0.25 to 100.00 µM were used to build the calibration curves. The linearity of the method was determined at nine concentration levels. The data are shown in Table 2 and Table 3. The relationship between APAP concentration and the peak areas was described by the linear regression equations: y = 16106*x + 3579.8 (R2=1, in DMEM medium) and y = 16397*x + 2475.1 (R2=1, in Williams medium), in which “x” is APAP nominal concentration in µM and “y” is the chromatogram peak area of APAP in AU. At the upper limit of quantification (i.e., 100.00 µM), the percentage deviation and the inter-run variability values were less than 2.50%. The accuracy and the precision for nine concentration levels, excluding the 0.50 µM (LLOQ), were within an acceptable range with DEV and C.V. values less than 7.00% (Table 2 and Table 3).
The analytical method inter- and intra-run accuracy and precision, at four tested concentrations, fell within the generally accepted criteria for bioanalytical assays. The reproducibility of the method was evaluated by analyzing replicates of APAP quality control samples of 0.50 (LLOQ), 4.50, 45.00 and 90.00 µM. The intra-run and inter-run average results are reported in Table 4. The accuracy and precision of the assay are demonstrated by DEV values ≤ 15.00% and by C.V. values ≤ 7.00%, respectively.
LOD was determined as the sample whose signal-to-noise ratio (S/N) was just greater than 3 and corresponded to a 0.25 µM APAP. On the other hand, the LLOQ, estimated with 0.50 µM APAP samples, displayed S/N ratio equal to 10. Furthermore, we found accuracy values (DEV%) ranging within ≤ 19.00% of the nominal concentration values. The intra- and inter-run variabilities were demonstrated by C.V. ≤ 18.77%, as shown in Table 2, Table 3 and Table 4)29,30.
The APAP PK analyses were performed in three different 2-OC MPS assemblies: 1) Intestine 2-OC, containing the intestine equivalent only; 2) Liver 2-OC, containing the liver spheroids only and 3) Intestine/Liver 2-OC with both intestinal barrier and liver spheroids.
For absorption studies, the oral route was mimicked by the administration of 12 µM APAP on the intestine equivalent apical side. APAP concentrations were measured by HPLC/UV, in the medium samples, collected from apical and basolateral intestinal equivalents sides, in both static and dynamic conditions. The APAP kinetics in the medium collected from the apical and basolateral sides demonstrated that the intestine model was able to absorb the APAP. There was a progressive APAP concentration decrease in the apical side (Figure 5A) concomitantly to APAP concentration increase at the intestinal basolateral side (Figure 5B). The maximum concentrations (Cmax) in the medium was around 2 µM for both static and dynamic conditions, after 12 h of the administration (Tmax).
For metabolism studies, the intravenous administration was mimicked by the application of 2 µM APAP in the medium of the liver compartment. The APAP concentration kinetics in the media under both static and dynamic conditions indicated that only at the dynamic conditions the decreases in the APAP concentration could be detected, reaching 0.87 µM APAP 12 h after 2 µM APAP administration (T1/2 = 12 h). The liver equivalents showed minimal metabolic efficiency under static conditions (Figure 5C). The APAP concentration reached 1.7 µM 12 h after APAP administration. The integrated, systemic like APAP absorption and metabolism evaluation was performed in the Intestine/Liver 2-OC model. The APAP was administered over the apical side of the intestine equivalent, emulating the oral route. Medium samples were collected from both Intestinal sides and also from the liver compartment. Figure 5D shows the progressive decay of the APAP concentration at the apical side in both static and dynamic conditions.
Figure 5E shows distinguishable absorption and metabolism phases. The flow also impacted in the intestinal absorption. The APAP Cmax in the medium changed from 2 µM on the “Intestine 2-OC” (Figure 5B) assembly to 1.7 µM for the dynamic “Intestine/Liver 2-OC” (Figure 5E). Figure 5F shows a direct comparison between the concentration–time profile of APAP in our dynamic “Intestine/Liver 2-OC” microphysiological system (red curve and y axis) and a representative profile obtained in humans after a single oral dose of 1000 mg (black curve and y axis).
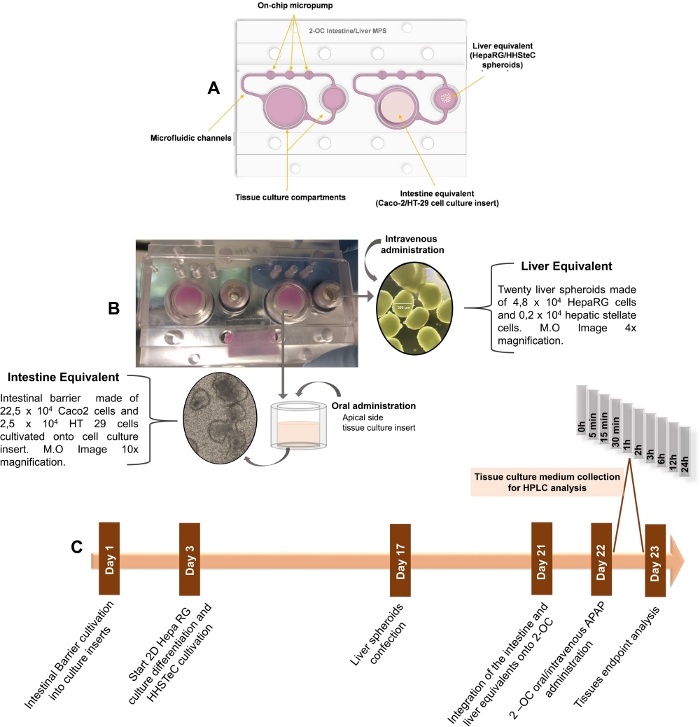
Figure 1: Schematic step compilation for PK studies in the 2-OC MPS. A) Schematic drawing of the 2-OC MPS, showing the intestinal and hepatic human tissue equivalents in bottom-up view. B) 2-OC MPS photograph with the intestinal and liver equivalents integrated into the device in bottom-up view, with representative optical microscopy images. C) Timeline of tissue equivalents preparation, APAP treatment, and culture medium collections phases for organoid manufacturing, and pharmacokinetic and toxicological assessments. This figure has been modified from Marin et al. Acetaminophen absorption and metabolism in an intestine/liver microphysiological system. Chem Biol Interact. 299, 59-76 (2019). Please click here to view a larger version of this figure.
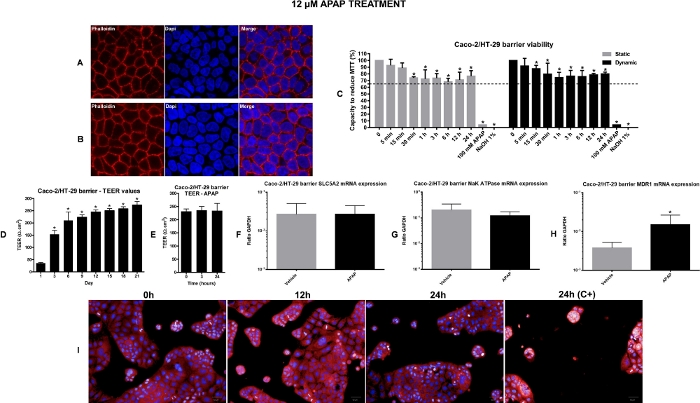
Figure 2: Viability and toxicological assessment of the intestine equivalents. A) Representative confocal fluorescence microscopy images of non-treated Caco-2/HT-29 cells stained with cell nuclei and actin filaments fluorescent dye (DAPI and Phalloidin respectively); 63x Magnification, zoom 2.6. B) Representative confocal fluorescence microscopy images of Caco-2/HT-29 cells treated with 12 µM APAP for 24 h, stained with nuclei and actin filaments fluorescent dye (DAPI and Phalloidin respectively); 63x Magnification, zoom 2.6. C) Intestine equivalents viability evaluation by MTT assay in both static and dynamic conditions. The values represented by the bars in the graph are percent calculated relative to vehicle control (time-point named as 0)*P<0.05 0 vs treatment. D) TEER evolution during the 21 days of differentiation. *P<0.05 day 1 vs other days. E) TEER values after APAP administration into the Intestine 2-OC MPS under dynamic conditions. Gene expression in intestines equivalents. Absorption potential of the intestine barrier and possible effects of 12 µM APAP for 24 h under dynamic condition was verified by SLC5A1 (F), Na-K-ATPase (G) and MDR1 (H) expression. Values represent the mean ± SEM of three independent experiments. The result is expressed as a ratio to housekeeping GAPDH. *P<0.05 vehicle vs APAP. I) Operetta image-based HCA performed by the Columbus® 2.4.0. software. Representative images of 2D intestine co-culture in different time points after 12 µM APAP treatment. Negative controls were medium (0 h) or vehicle (0.5% ethanol). Positive controls shown here is the 100 mM APAP. This figure has been modified from Marin et al. Acetaminophen absorption and metabolism in an intestine/liver microphysiological system. Chem Biol Interact. 299, 59-76 (2019). Please click here to view a larger version of this figure.
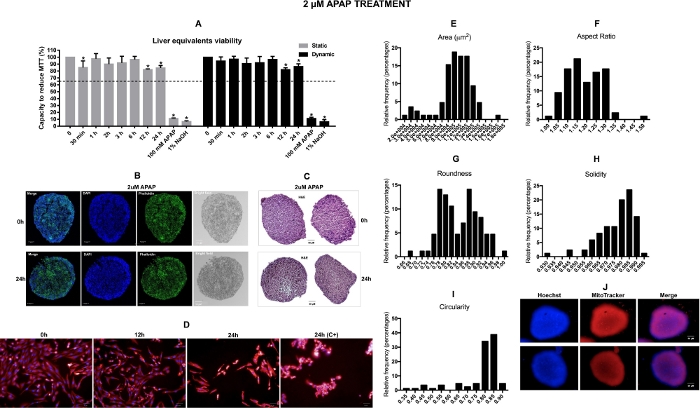
Figure 3: Viability and toxicological assessment of the liver equivalents. A) Liver equivalents viability evaluation by MTT assay in both static and dynamic conditions. The values represented by the bars in the graph are percent calculated relative to vehicle control (time-point named as 0) *P<0.05 0 vs treatment. B) Representatives confocal images captured from the vehicle and 2 µM APAP 24 h treated liver spheroids from an inner section. C) Representatives H&E (hematoxylin and eosin staining) images captured from the vehicle and 2 µM APAP 24 h treated liver spheroids from an inner section. Scale bar = 50 µm. D) Representative images of 2D liver co-culture in different time points after 2 µM APAP treatment. Samples treated with vehicle and with 2 µM APAP were considered in these analyses. The fluorophore dye mixture includes Hoechst for nuclear staining and Mitotracker Deep Red for mitochondria mass staining. Negative controls were medium (0 h) or vehicle (0.5% ethanol). Positive controls were 100 mM APAP. Measurements of whole spheroids images captured were performed using Fiji software. E) Frequency distributions of the area F) aspect ratio, G) roundness, H) solidity, I) circularity. N = 85. *p < 0,05. J) Representative images of 3D liver spheroids acquired by the Operetta using the LWD 10x objective. This figure has been modified from Marin et al. Acetaminophen absorption and metabolism in an intestine/liver microphysiological system. Chem Biol Interact. 299, 59-76 (2019). Please click here to view a larger version of this figure.
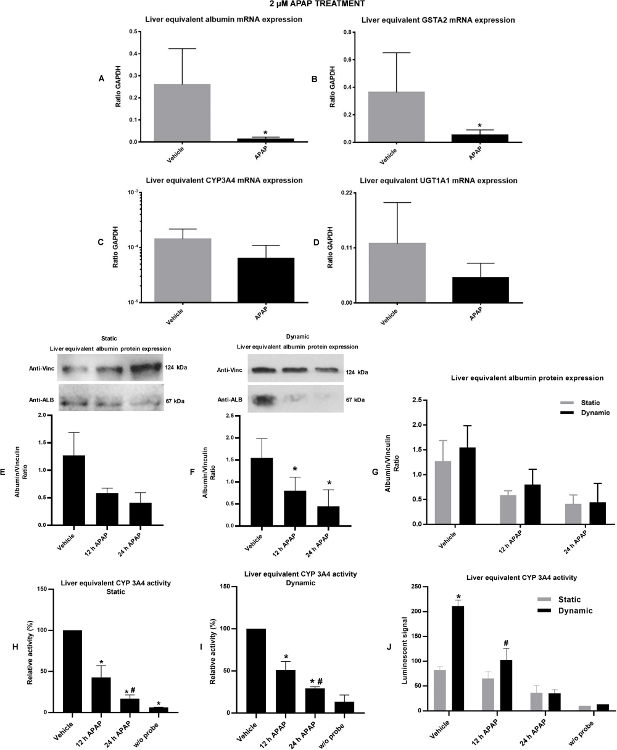
Figure 4: Liver viability/functionality and possible effects of 2 µM APAP under static and dynamic conditions over it were verified by gene and protein expression and by enzymatic activity. A) albumin gene expression. B) GSTA2 gene expression. Liver capability to perform phase I and phase II metabolism and possible effects of 2 µM APAP for 24 h under dynamic condition over it were verified by gene expression of CYP3A4 (C) and by UGT1A1 (D) respectively. E) Total albumin protein expression under static condition. F) Total albumin protein expression under dynamic conditions. condition. G) Comparative graph illustrating the difference in total albumin expression in liver equivalents cultivated and treated under static or dynamic conditions. H) CYP 3A4 in vitro enzymatic activity under static conditions. I) CYP 3A4 in vitro enzymatic activity under dynamic conditions. J) Comparative graph illustrating the difference in CYP 3A4 activity in liver equivalents cultivated and treated under static or dynamic conditions. Values represent the mean ± SEM of three independent experiments. The data of gene expression is expressed as a ratio to housekeeping GAPDH. The data of protein expression is expressed as a ratio to vinculin protein. *P<0.05 vehicle vs APAP treatment. This figure has been modified from Marin et al. Acetaminophen absorption and metabolism in an intestine/liver microphysiological system. Chem Biol Interact. 299, 59-76 (2019). Please click here to view a larger version of this figure.
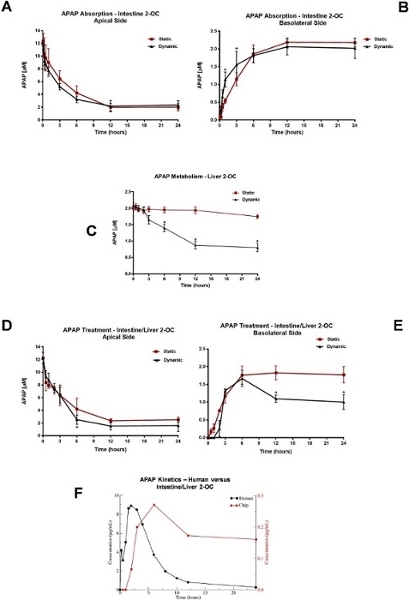
Figure 5: Analyzes of APAP pharmacokinetics in 2-OC MPS. APAP absorption profile after 12 µM APAP administration at the apical side of the Intestine 2-OC MPS preparation. The intestinal barrier was made of a coculture of Caco-2/HT-29 cell lines (A) Static and dynamic APAP concentrations in the medium from the apical side (representing the human intestinal lumen side). (B) Static and dynamic APAP concentrations in the medium from the basolateral side (representing the human intestinal bloodstream side). *P<0.05 static vs dynamic conditions. C) APAP metabolism profile in the Liver 2-OC MPS by HepaRG/HHSTeC liver spheroids. Comparison of static and dynamic conditions after a 2 µM APAP administration into the medium. *P<0.05 0h vs 6 h, 12 h, and 24 h APAP treatment. APAP absorption and metabolism profile after 12 µM administration on the intestinal barrier apical side of the Intestine/Liver 2-OC MPS preparation. This emulates the oral route. The intestinal barrier was made of Caco-2/HT-29 cell lines and the liver equivalent made of spheroids of HepaRG/HHSTeC cell lines. (D) Intestinal/Liver 2-OC APAP concentrations in the apical side of the intestinal barrier under static and dynamic conditions. (E) Intestinal/Liver 2-OC APAP concentrations in the medium under static and dynamic conditions. *P<0.05 static vs dynamic conditions. (F) Comparison between the concentration–time profile of APAP in our microphysiological system (red curve and y axis) and a representative profile obtained in humans after a single oral dose of 1000 mg (black curve and y axis). Data was extracted from plots using WebPlotDigitizer 4.2 (https://automeris.io/WebPlotDigitizer). This figure has been modified from Marin et al. Acetaminophen absorption and metabolism in an intestine/liver microphysiological system. Chem Biol Interact. 299, 59-76 (2019). Please click here to view a larger version of this figure.
| HPLC system | Waters Alliance 2695 (Milford, MA, USA), equipped with quaternary pump, sample manager and degasser | ||
| Detector | Waters 2996 Uv-Vis set in 210-400 nm range | ||
| System control, data acquisition, and processing | Waters Empower 2002 chromatography software | ||
| Column | Reversed-phase Luna C18 | ||
| (150 x 4.6 mm I.D.; 5mm particle size) | |||
| Phenomenex | |||
| Guard Column | Reversed-phase Luna C18 (4 x 3 mm) | ||
| Phenomenex | |||
| Mobile phase | Solvent A- Acetonitrile | ||
| Solvent B- 0.10 M ammonium acetate, pH 6.8 | |||
| Isocratic conditions | Time | A (%) | B (%) |
| (min.) | |||
| 15 | 5 | 95 | |
| Flow | 1.0 mL/min | ||
| Injection volume | 25 μL | ||
| Temperature | 25 °C | ||
| APAP Detection | UV @ 243 and 254 nm | ||
| Run time | 15 minutes | ||
Table 1: Conditions and parameters to be used for HPLC-UV analyses of APAP in culture medium matrices.
| Nominal concentration | Calculated APAP concentration (µM) | Average (µM) | S.D.b | C.V. | DEV | |||||
| (µM) | (Triplicate of each concentration) | (µM) | (%)c | (%)d | ||||||
| Assay number | 1 | 2 | 3 | 4 | 5 | 6 | ||||
| 0.25 (LOD) | 0.02 | 0.08 | 0.21 | 0.14 | 0.08 | 0.27 | 0.13 | 0.11 | 84.96 | + 46.05 |
| 0.50 (LLOQ) | 0.31 | 0.36 | 0.29 | 0.47 | 0.42 | 0.65 | 0.41 | 0.05 | 12.72 | + 17.08 |
| 1.00 | 0.87 | 0.87 | 0.80 | 1.04 | 1.02 | 1.01 | 0.93 | 0.04 | 3.76 | + 6.65 |
| 2.50 | 2.44 | 2.61 | 2.52 | 2.42 | 2.56 | 2.54 | 2.52 | 0.04 | 1.54 | -0.62 |
| 5.00 | 5.02 | 4.99 | 5.06 | 5.01 | 5.01 | 5.05 | 5.02 | 0.09 | 1.88 | -0.45 |
| 10.00 | 10.21 | 10.13 | 9.96 | 10.25 | 10.08 | 10.21 | 10.14 | 0.10 | 0.97 | -1.41 |
| 25.00 | 25.33 | 25.28 | 25.20 | 25.13 | 25.14 | 24.92 | 25.17 | 0.36 | 1.45 | -0.67 |
| 50.00 | 50.30 | 50.04 | 50.51 | 49.70 | 49.98 | 49.19 | 49.95 | 0.86 | 1.71 | +0.09 |
| 100.00 | 99.75 | 99.90 | 99.70 | 100.09 | 99.97 | 100.40 | 99.97 | 0.69 | 0.69 | +0.03 |
| R2 | 1.00 | 1.00 | 0.9999 | 1.00 | 1.00 | 0.9999 | ||||
Table 2: Inter-run variation – accuracy, precision, and linearity of standard curve samples prepared in a mixture of DMEM medium with 0.10 M ammonium acetate buffer (1:1, v/v) from six separate assays.a
aA linear curve was fitted to the data for a response (APAP) versus theoretical concentration as described in Experimental. The calculated concentration was derived from reading the response for each standard sample against the calibration curve. Each entry (assay 1-6) corresponds to the average value of triplicate analysis.
bSD= Standard deviation.
cC.V. (coefficient of variation. precision).
dAccuracy (DEV %) = the deviation of the calculated concentration from the nominal value.
This figure has been modified from Marin et al. Acetaminophen absorption and metabolism in an intestine/liver microphysiological system. Chem Biol Interact. 299, 59-76 (2019).
| Nominal concentration | Calculated APAP concentration (µM) | Average | S.D.b | C.V. | DEV | ||||
| (µM) | (Triplicate of each concentration) | (µM) | (µM) | (%)c | (%)d | ||||
| Assay number | 1 | 2 | 3 | 4 | 5 | ||||
| 0.25 (LOD) | 0.34 | 0.12 | 0.30 | 0.16 | 0.00 | 0.18 | 0.08 | 45.46 | +26.03 |
| 0.50 (LLOQ) | 0.36 | 0.44 | 0.49 | 0.43 | 0.40 | 0.43 | 0.02 | 5.84 | +14.97 |
| 1.00 | 0.87 | 0.98 | 1.04 | 0.94 | 0.83 | 0.93 | 0.04 | 4.67 | +6.85 |
| 2.50 | 2.41 | 2.46 | 2.49 | 2.52 | 2.43 | 2.46 | 0.06 | 2.39 | +1.56 |
| 5.00 | 5.00 | 4.99 | 5.12 | 5.10 | 5.14 | 5.07 | 0.15 | 3.00 | -1.38 |
| 10.00 | 10.08 | 10.01 | 9.93 | 10.10 | 10.29 | 10.08 | 0.18 | 1.80 | -0.81 |
| 25.00 | 25.14 | 25.18 | 24.96 | 25.32 | 25.35 | 25.19 | 0.45 | 1.78 | -0.76 |
| 50.00 | 50.19 | 50.23 | 49.83 | 49.56 | 50.17 | 49.99 | 0.87 | 1.75 | +0.01 |
| 100.00 | 99.87 | 99.84 | 100.10 | 100.13 | 99.80 | 99.95 | 2.12 | 2.12 | +0.05 |
| R2 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | ||||
Table 3: Inter-run variation – accuracy, precision, and linearity of standard curve samples prepared in a mixture of Williams medium with 0.10 M ammonium acetate buffer (1:1, v/v) from six separate assays.a
aA linear curve was fitted to the data for a response (APAP) versus theoretical concentration as described in Experimental. The calculated concentration was derived from reading the response for each standard sample against a calibration curve. Each entry (assay 1-5) corresponds to the average value of triplicate analysis.
bSD= Standard deviation.
cC.V. (coefficient of variation. precision).
dAccuracy (DEV %) = the deviation of the calculated concentration from the nominal value.
This figure has been modified from Marin et al. Acetaminophen absorption and metabolism in an intestine/liver microphysiological system. Chem Biol Interact. 299, 59-76 (2019).
| Williams medium | Nominal concentration | Measured concentration | S.D. | C.V. | DEV |
| (µM) | (µM) | (µM) | (%) | (%) | |
| Intra-run (n=3) | 0.50 (LLOQ) | 0.49 | 0.08 | 15.55 | +1.77 |
| 4.50 | 4.59 | 0.23 | 5.10 | -2.06 | |
| 45.00 | 41.23 | 0.76 | 1.85 | +8.37 | |
| 90.00 | 82.29 | 1.75 | 2.13 | +8.57 | |
| Inter-run (n=15) | 0.50 (LLOQ) | 0.43 | 0.05 | 10.99 | +14.97 |
| 4.50 | 4.37 | 0.19 | 4.42 | +2.99 | |
| 45.00 | 42.35 | 0.82 | 1.93 | +5.88 | |
| 90.00 | 85.22 | 2.25 | 2.65 | +5.31 | |
| DMEM medium | Nominal concentration | Measured concentration | S.D. | C.V. | DEV |
| (µM) | (µM) | (µM) | (%) | (%) | |
| Intra-run (n=3) | 0.50 (LLOQ) | 0.47 | 0.09 | 18.77 | +6.35 |
| 4.50 | 4.45 | 0.30 | 6.63 | +1.04 | |
| 45.00 | 44.24 | 1.59 | 3.58 | +1.69 | |
| 90.00 | 86.40 | 4.09 | 4.73 | +4.00 | |
| Inter-run (n=12) | 0.50 (LLOQ) | 0.46 | 0.08 | 17.81 | +7.75 |
| 4.50 | 5.16 | 0.27 | 5.31 | -14.68 | |
| 45.00 | 48.99 | 2.10 | 4.29 | -8.86 | |
| 90.00 | 96.18 | 4.47 | 4.65 | -6.86 | |
Table 4: Intra- and inter-run precision and accuracy for APAP in quality control samples.a
aThe data are shown as averages. SD (standard deviation). C.V. (coefficient of variation. precision) and accuracy (percent deviation. DEV%).
This figure has been modified from Marin et al. Acetaminophen absorption and metabolism in an intestine/liver microphysiological system. Chem Biol Interact. 299, 59-76 (2019).
| Primer (5’ → 3’) | |||||
| Tissue | Gene | Forward | Reverse | ||
| Intestine | SGLT1/SLC5A1 | gAgCCCAgCAACTgTCCCAC | CAggCTCCAACACAgACggT | ||
| NA-K-ATPase | ACCgCCCAgAAATCCCAAAAC | CAgCggTCATCCCAgTCC | |||
| MDR1 | TggATgTTTCCggTTTggAg | TgTgggCTgCTgATATTTTgg | |||
| Liver | Albumin | TgCAAggCTgATAAggAg | TTTAgACAgggTgTTggCTTTACAC | ||
| GSTA2 | CTgAggAACAAgATgCCAAgC | AgCAgAgggAAggCTggAAATAAg | |||
| CPY3A4 | ggAAggTggACCCAgAAACTgC | TTACggTgCCATCCCTTgAC | |||
| UGT1A1 | ATgCAAAgCgCATggAgAC | ggTCCTTgTgAAggCTggAg | |||
Table 5: Real-time qPCR primers to evaluate gene transcription at mRNA level in the 2-OC cultures for intestine and liver tissues
