Herein, we describe a method for DNA isolation from intestinal tissue and blood, preparation of libraries for NGS, and basic steps of a sequencing run for immune repertoire sequencing. The run will generate fastq files, which can be further converted to fasta files for use in the international ImMunoGeneTics (IMGT)/HighV-QUEST platform. This HTS performs and manages many analyses of tens of thousands of rearranged TCRβ and IGH sequences, at the nucleotide level15. IMGT/HighV-QUEST enables analysis of different TCRs and IGH repertoires in both health and disease. This can lead to identification of new "disease-specific" clones, analysis of clonal expansion and diversity parameters, delineation of differential V(D)J usage, analysis of somatic hyper-mutations, and more. The IMGT/HighV-QUEST provides CSV files, which contain specific sequences and their abundance. Using genomic DNA as a starting material yields sequence numbers that are representative of cell numbers. Thus, if original DNA quantity is equal, percentage of T cells in a given sample can also be calculated.
We include a basic analysis from representative, autologous blood and rectal samples of a patient with IL10 receptor deficiency and history of severe infantile-onset IBD, resulting from a deleterious IL10RA mutation. Samples were assayed for both TCRβ and IGH repertoire. The intestinal TCRβ sample yielded a total 12,450 sequences, of which 9,050 sequences were unique. The blood TCRβ sample had a total 54,880 sequences, of which 35,110 were unique. In the intestinal IGH sample, a total of 49,070 sequences were obtained, of which 23,670 were unique. In the blood IGH sample, a total of 13,710 sequences were obtained, of which 13,540 were unique. All clones can be identified by their unique sequence both at the nucleotide or amino acid level. These sequences can be compared between different patients, in search for shared clones, or in the same patient between different anatomical sites (e.g., blood vs. intestine). We present for each of the samples the 5 most frequent clones (Table 1).
To quantify the degree of clonal expansion different indices can be used, including Shannon’s H, Gini-Simpson, entropy and clonality. As an example, Shannon's H, which takes into account the number of unique sequences (richness of the repertoire) and how evenly they are distributed was found to be decreased in the patient's intestinal IGH vs. blood (8.3 vs. 9.5), suggesting clonal expansion of B cells in the inflamed gut.
For a broad overview of the repertoire, Treemap images (www.treemap.com) were generated (Figure 2). Each colored square represents a different clone, and the size correlates with its frequency. These Treemap images demonstrate clonal expansion in the intestinal IGH repertoire compared with the blood. In contrast, in the TCRβ repertoire, marked clonal expansion is observed in the blood, in comparison to autologous intestine.
At the gene level, NGS provides information regarding V- D- and J- usage at the level of either gene, family, or allele, as shown in Table 1. Moreover, specific V(D)J combinations can be inferred from the data for TCRβ and IGH repertoires (Figure 3A,B), which can reveal differential gene usage patterns in various conditions. Biophysical properties of the CDR3 region such as length (Table 1) or hydrophobicity can also be analyzed. Importantly, CDR3 length distribution is altered in different immune-mediated disorders10,16,17. For example, in IL10R deficient patients, blood-derived T cells, but not B cells, have shorter CDR3 length, and differential hydrophobicity, in comparison to healthy controls7.
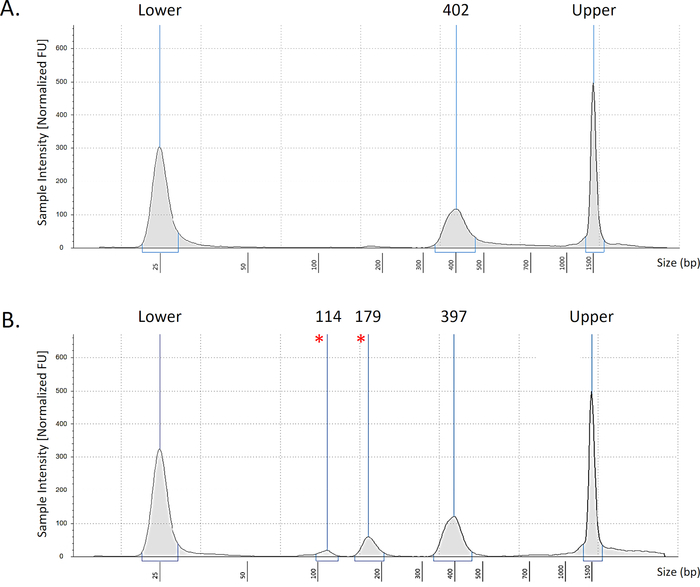
Figure 1: Representative bioanalyzer data. Representative images of intestinal TCRβ samples showing optimal results (A) with the desired peak at 400 bp. An example of a low-quality library (B) with additional peaks at 179bp and 114bp (marked *). Peaks seen at ends are upper and lower markers of the electrophoresis strip. Please click here to view a larger version of this figure.
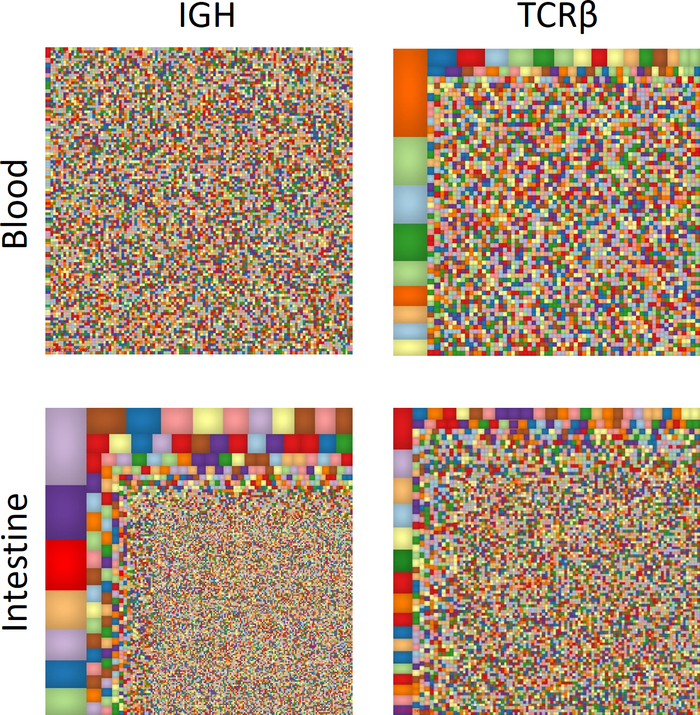
Figure 2: Overview of T and B cell immune repertoire. Treemap diagrams of TCRβ and IGH blood and intestinal samples. Each colored square represents a different clone, and the size correlates with its frequency. Please click here to view a larger version of this figure.
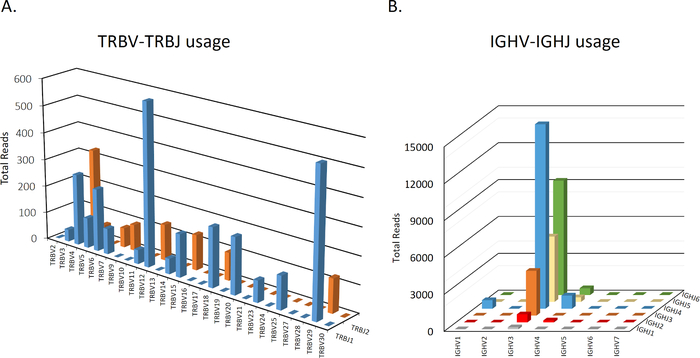
Figure 3: Representative graphs of TCRβ and IGH V-J usage. A representative intestinal sample depicting total number of sequences for each V-J combination in TCRβ (A) or IGH (B). Data is not shown for D usage. Please click here to view a larger version of this figure.
