Para comprender las funciones de los ARN catalíticos y no codificantes implicadas en la regulación del corte y empalme, traducción, replicación del virus y el cáncer, se requiere un conocimiento detallado de la estructura del ARN 1,2. Desafortunadamente, la predicción exacta de ARN plegado presenta un reto formidable. Agentes de sondeo clásicas sufren de muchas desventajas tales como la toxicidad, la cobertura incompleta de nucleótidos y / o un caudal limitado a 100-150 nucleótidos por experimento. Algoritmos de predicción de estructura secundaria sin ayuda son igualmente desventajoso, debido a las inexactitudes que resultan de su incapacidad para distinguir efectivamente entre estructuras energéticamente similares. Grandes de ARN, en particular, también son a menudo refractario a los métodos de determinación de la estructura 3D, tales como la cristalografía de rayos X y resonancia magnética nuclear (RMN), debido a su flexibilidad conformacional y grandes cantidades de muestras muy puras requeridas para estas técnicas.
HFORMA igh rendimiento resuelve muchos de estos problemas al proporcionar un enfoque eficaz, sencilla de sondeo de las grandes estructuras de ARN a una resolución de un solo nucleótido. Por otra parte, los reactivos utilizados para la forma son seguros, fáciles de manejar y, en contraste con la mayoría de otros productos químicos de sondeo reactivos, reaccionan con los cuatro ribonucleótidos. Estos reactivos también pueden penetrar en las membranas celulares, por lo que es posible para sondear RNAs en su contexto in vivo (s) 3. Originalmente desarrollado en las Semanas de laboratorio 4, la forma ha sido utilizado para analizar una amplia variedad de ARN, el ejemplo más notable es la determinación de la estructura secundaria completa del ~ 9 kb VIH-1 ARN del genoma 5. Otros logros notables incluyen SHAPE utilizando elucidación de las estructuras de los viroides infecciosas 6, ARNs no codificantes largas humanos 7, ribosomas de levadura 8 y 9 riboswitches así como para identificar los sitios de unión de proteínas de virión asociada a VIH-1 RNA 3. While las variaciones originales y de alto rendimiento del protocolo SHAPE se han publicado en otros lugares 10 a 12, el presente trabajo ofrece una descripción detallada de la determinación de la estructura secundaria del ARN por la forma de alto rendimiento utilizando oligonucleótidos fluorescentes, la Beckman Coulter CEQ 8000 Genetic Analyzer y software SHAPEfinder y RNAStructure (v5.3). Detalles técnicos inéditas y consejos de solución de problemas también se incluyen.
Las variaciones de la forma
La esencia de la forma y sus variaciones es la exposición de ARN en solución acuosa a anhídridos electrófilos que acilar selectivamente grupos ribosa 2'-hidroxilo (2'-OH), produciendo aductos voluminosos en los sitios de modificación. Esta reacción química sirve como un medio de interrogar a la dinámica estructural de ARN locales, como los nucleótidos de cadena simple son más propensas a adoptar conformaciones favorables para ataque electrofílico por estos reactivos, mientras que la base emparejado o arquitectónicamente Constrnucleótidos objetivó son menos o no reactivo 10. Los sitios de formación de aductos se detectan mediante transcripción inversa a partir de la iniciación fluorescentemente o radiomarcado cebadores hibridados a un sitio específico en el ARN modificado (el "(+)" reacción de extensión del cebador). Cuando la transcriptasa inversa (RT) no atravesar los ribonucleótidos acilados, un grupo de productos de ADNc se produce cuyas longitudes coinciden con los sitios de modificación. Un control, "(-)" cebador de extensión reacción utilizando ARN que no ha sido expuestos a reactivo también se realiza de modo que la terminación prematura de la síntesis de ADN (es decir, "se detiene") debido a la estructura del ARN, no específica de ARN de hebra rotura, etc, mayo. distinguirse de deteniéndose producido por modificación química. Por último, dos reacciones de secuenciación didesoxi-inician a partir de los mismos cebadores se utilizan como marcadores para correlacionar nucleótidos reactivos con la secuencia primaria de ARN después de la electroforesis.
En la solicitud original de SHAPE, el mismo 32-P marcado en el extremo cebador se utiliza para los (+), -, y dos reacciones de secuenciación (). Los productos de estas reacciones se cargan en pocillos adyacentes en una placa de gel de poliacrilamida al 5-8%, y se fraccionó por electroforesis en gel desnaturalizante de poliacrilamida (PAGE; Figura 1). El análisis cuantitativo de las imágenes de gel producidas por la forma convencional se puede realizar utilizando SAFA, un software de análisis de la huella de semi-automatizado 13.
Por el contrario, la forma de alto rendimiento emplea cebadores marcados con fluorescencia y electroforesis capilar automatizado. Específicamente, para cada región de ARN bajo investigación, un conjunto de cuatro cebadores de ADN que tienen una secuencia común pero diferentes 5 'etiquetas fluorescentes deben ser sintetizados o comprados. Estos oligonucleótidos marcados de manera diferente-sirven para dos reacciones principales forma y dos reacciones de secuenciación, los productos de los cuales se agrupan y se fraccionó / detectaron por electroforesis capilar automatizado (CE). WherEAS el perfil de reactividad de los nt 100-150 de ARN se puede obtener a partir de un conjunto de cuatro reacciones utilizando el enfoque original, la forma de alto rendimiento permite la resolución de 300-600 nt a partir de una sola muestra agrupada 3. Hasta 8 conjuntos de reacciones puede ser fraccionado simultáneamente, mientras que tanto como 96 muestras pueden preparado para el fraccionamiento en el transcurso de 12 ejecuciones consecutivas de la CE (Figura 2). Por otra parte, el software SHAPEfinder, desarrollado para procesar y analizar los datos que se desprenden del CEQ y otros analizadores genéticos, es más automatizado y requiere mucho menos intervención del usuario de SAFA 13 u otros paquetes de gel de análisis.
Más avanzadas metodologías de alto rendimiento han surgido recientemente tales como PARS (análisis paralelo de la estructura del ARN) 14 y Frag-Seq (fragmento de secuenciación) 15, que utilizan enzimas específicos de la estructura en lugar de reactivos de alquilación en conjunción con técnicas de secuenciación de próxima generación para obtener information sobre la estructura del ARN. A pesar del atractivo de estas técnicas, las muchas limitaciones inherentes a la nucleasa de sondeo siguen siendo 16. Estos problemas pueden ser evitados en la secuenciación FORMA (SHAPE-Seq) 17 protocolo, donde la secuenciación de próxima generación está precedida por modificación química y la transcripción inversa del ARN de una manera similar a la realizada en la forma convencional. Si bien estos métodos pueden representar el futuro de la determinación de la estructura de ARN, es importante recordar que la secuenciación de próxima generación es muy caro, y sigue sin estar disponible para muchos laboratorios.
Análisis de datos SHAPE
Los datos producidos en el analizador genético se presentan en la forma de un electroferograma, en el que la intensidad de fluorescencia de la muestra (s) que fluye a través del detector capilar se representa en función de un índice de tiempo de migración. Esta parcela tiene la forma de las huellas superpuestas, según el canal cuatro de fluorescencias utiliza para detectar los diferentes fluoróforos, y donde cada traza se compone de los picos correspondientes a los productos de ADNc o secuenciación individuales. Datos electroferograma se exporta desde el analizador genético como un archivo de texto delimitado por tabuladores y se importan en la transformación ShapeFinder y software de análisis 18.
ShapeFinder se utiliza inicialmente para llevar a cabo una serie de transformaciones matemáticas en los datos para asegurar que los tiempos de migración y volúmenes pico reflejan con precisión las identidades y cantidades de los productos de reacción, respectivamente. Los picos son entonces alineados e integrados, y los resultados se tabulan junto con la secuencia de ARN primario. Un "perfil de reactividad" para el segmento pertinente de ARN se obtiene restando los valores de control a partir de la (+) valores asociados con cada ARN de nucleótidos, y la normalización de los datos tal como se describe a continuación. Este perfil se importa en RNAstructure (v5.3) Software de 19,20, lo que convierte la val reactividad normalizadaUES en limitaciones pseudo-energía que se incorporan en el algoritmo de plegado estructura secundaria del ARN. La combinación química de sondeo y plegado algoritmos de esta manera mejora significativamente la exactitud de la predicción de la estructura en comparación con cualquiera de los métodos solos 12,21. La salida de RNAstructure (v5.3) incluye imágenes de la energía más baja ARN estructuras secundarias con código de colores con el perfil FORMA reactividad (s), así como las mismas estructuras en Pruebas notación de punto-soporte. Este último puede posteriormente ser exportado al software dedicado a la representación gráfica de la estructura secundaria del ARN tales como Varna PseudoViewer 22 y 23.
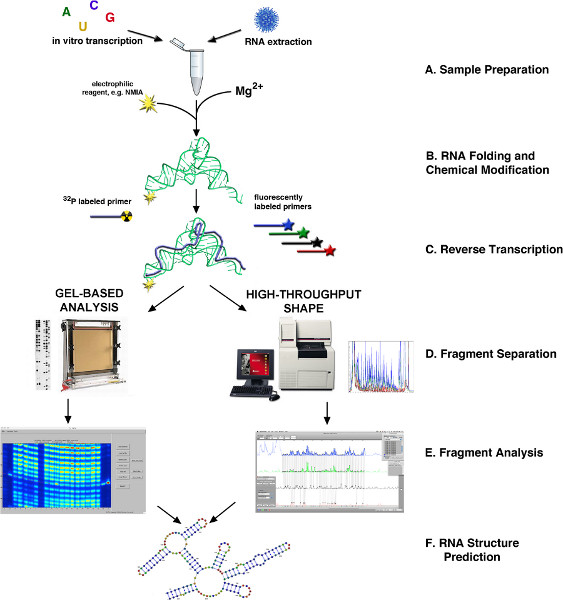
Figura 1. Organigrama de la estructura de la determinación de ARN a través de SHAPE 4,10. (A) RNA may se obtiene a partir de muestras biológicas o por transcripción in vitro. (B) Dependiendo de la fuente, el ARN se pliega o se procesa y se modificó con el reactivo FORMA lo contrario. (C) La transcripción inversa usando cebadores marcados radiactivamente o fluorescentemente. (D) son productos de ADNc fraccionado ya sea a través de electroforesis capilar a base de gel o de poca altura. (E) Análisis de fragmentos. (F) la predicción de estructura de ARN. Haz clic aquí para ver más grande la figura.
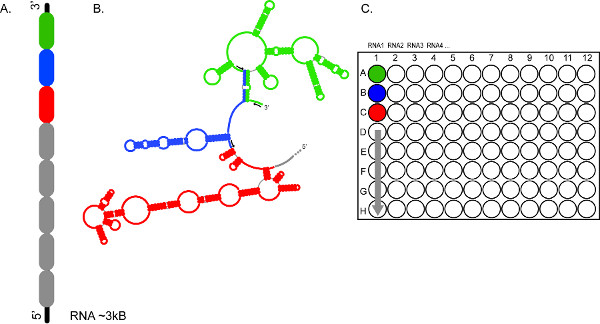
Figura 2. El carácter de alto rendimiento de FORMA basados en CE permite el análisis rápido de múltiples ARN, y / o múltiples segmentos de los mismos ARN. (A) </strong> Representa cómo una RNA puede ser dividido en secciones 300-600 nt (código de color en verde, azul y rojo) (B) Las secciones del ARN se probaron de forma independiente utilizando diferentes conjuntos de cebadores fluorescentes (flechas negras) (C) juegos de reacciones se combinaron y se cargaron en los pocillos A1, B1, C1, etc, respectivamente, proporcionando una cobertura completa para el ~ 3 kb ARN1. Productos de reacción de RNAs 2, 3, 4, etc pueden prepararse de forma similar para el fraccionamiento de carreras electroforéticas consecutivos. Haz clic aquí para ver más grande la figura.
