Combining X-Ray Crystallography with Small Angle X-Ray Scattering to Model Unstructured Regions of Nsa1 from S. Cerevisiae
Summary
This method describes the cloning, expression, and purification of recombinant Nsa1 for structural determination by X-ray crystallography and small-angle X-ray scattering (SAXS), and is applicable for the hybrid structural analysis of other proteins containing both ordered and disordered domains.
Abstract
Determination of the full-length structure of ribosome assembly factor Nsa1 from Saccharomyces cerevisiae (S. cerevisiae) is challenging because of the disordered and protease labile C-terminus of the protein. This manuscript describes the methods to purify recombinant Nsa1 from S. cerevisiae for structural analysis by both X-ray crystallography and SAXS. X-ray crystallography was utilized to solve the structure of the well-ordered N-terminal WD40 domain of Nsa1, and then SAXS was used to resolve the structure of the C-terminus of Nsa1 in solution. Solution scattering data was collected from full-length Nsa1 in solution. The theoretical scattering amplitudes were calculated from the high-resolution crystal structure of the WD40 domain, and then a combination of rigid body and ab initio modeling revealed the C-terminus of Nsa1. Through this hybrid approach the quaternary structure of the entire protein was reconstructed. The methods presented here should be generally applicable for the hybrid structural determination of other proteins composed of a mix of structured and unstructured domains.
Introduction
Ribosomes are large ribonucleoprotein machines that carry out the essential role of translating mRNA into proteins in all living cells. Ribosomes are composed of two subunits which are produced in a complex process termed ribosome biogenesis1,2,3,4. Eukaryotic ribosome assembly relies on the aid of hundreds of essential ribosomal assembly factors2,3,5. Nsa1 (Nop7 associated 1) is a eukaryotic ribosome assembly factor that is specifically required for the production of the large ribosomal subunit6, and is known as WD-repeat containing 74 (WDR74) in higher organisms7. WDR74 has been shown to be required for blastocyst formation in mice8and the WDR74 promoter is frequently mutated in cancer cells9. However, the function and precise mechanisms of Nsa1/WDR74 in ribosome assembly are still largely unknown. To begin to uncover the role of Nsa1/WDR74 during eukaryotic ribosome maturation, multiple structural analyses were performed, including X-ray crystallography and small angle X-ray scattering (SAXS)10.
X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, electron microscopy, and SAXS are all important techniques for studying macromolecular structure. Size, shape, availability, and stability of macromolecules influences the structural biology method for which a particular macromolecule will be best suited, however combining multiple techniques through a so-called "hybrid" approach is becoming an increasingly beneficial tool11. In particular X-ray crystallography and SAXS are powerful and complementary methods for structural determination of macromolecules12.
Crystallography provides high-resolution atomic structures ranging from small molecules to large cellular machinery such as the ribosome, and has led to numerous breakthroughs in the understanding of the biological functions of proteins and other macromolecules13. Furthermore, structure-based drug design harnesses the power of crystal structures for molecular docking by computational methods, adding a critical dimension to drug discovery and development14. Despite its broad applicability, flexible and disordered systems are challenging to assess by crystallography since crystal packing can be hindered or electron density maps may be incomplete or of poor quality. Conversely, SAXS is a solution-based and low-resolution structural approach capable of describing flexible systems ranging from disordered loops and termini to intrinsically disordered proteins12,15,16. Considering it is compatible with a broad range of particle sizes12, SAXS can work synergistically with crystallography to expand the range of biological questions that can be addressed by structural studies.
Nsa1 is suitable for a hybrid structural approach because it contains a well-structured WD40 domain followed by a functional, but flexible C-terminus which is not amenable to X-ray crystallography methods. Following is a protocol for the cloning, expression, and purification of S. cerevisiae Nsa1 for hybrid structural determination by X-ray crystallography and SAXS. This protocol can be adapted to study the structures of other proteins that are comprised of a combination of ordered and disordered regions.
Protocol
1. Recombinant Protein Production and Purification of Nsa 1
- Nsa1 Expression Plasmid Design and Cloning
- Obtain or purchase S. cerevisiae genomic DNA.
- PCR amplify the target sequences of Nsa1 (Nsa1FL, residues 1-463) and C-terminal truncated Nsa1 (Nsa1ΔC, residues 1-434) with appropriate primers using genomic DNA isolated from S. cerevisiae and a melting temperature of approximately 60 °C with an extension time of 1-2 min. The following primers were used to amplify Nsa1:
SC_Nsa1_FLFw:CGCCAAAGGCCTATGAGGTTACTAGTCAGCTGTGTGGATAG
SC_Nsa1_FLRv:AATGCAGCGGCCGCTCAAATTTTGCTTTTCTTACTGGCTTTAGAAGCGC
SC_Nsa1ΔcFw:GGGCGCCATGGGATCCATGAGGTTACTAGTCAGCTGTGTGG
SC_Nsa1ΔcRv: GATTCGAAAGCGGCCGCTTAAACCTTCCTTTTTTGCTTCCC - Subclone Nsa1 into the Escherichia coli (E. coli) expression vector pHMBP containing an N-terminal 6X-Histidine tag followed by the Maltose Binding Protein (MBP) and a Tobacco Etch Virus (TEV) protease site using standard cloning techniques17.
- Use DNA sequencing to verify that Nsa1 was cloned in frame with the N-terminal His-MBP tag.
- Nsa1 Protein Expression
- Transform the expression plasmid(s) into a suitable E. coli expression strain with a T7 promoter-based system. Plate the transformants on LB agar plates containing 100 µg/mL ampicillin and incubate the plate inverted overnight at 37 °C.
- Inoculate a 50 mL culture of LB with 100 µg/mL ampicillin from the transformation plate, and grow it overnight with shaking at 200 rpm at 37 °C.
- Inoculate 3 x 1000 mL of LB in Fernbach flasks with 100 µg/mL ampicillin with 15 mL of the overnight culture and grow it with shaking at 200 rpm at 37 °C.
NOTE: For protein structure solution, selenomethionyl (SeMet) incorporation can be achieved by growing cells in M9 minimal medium that is supplemented with SeMet and an amino acid mixture to inhibit methionine production prior to induction, as opposed to LB media18. - Induce expression of Nsa1 when the OD600 reaches ~0.8 by addition of isopropyl β-D-1 thiogalactopyranoside (IPTG) at a final concentration of 1 mM followed by incubation at 25 °C overnight with shaking at 200 rpm.
- Harvest cells by centrifugation at 4 °C for 15 min at 5,050 x g.
NOTE: Cells can be stored long-term at -80 °C or used immediately for protein purification.
- Nsa1 Protein Purification
- Resuspend the cells in 25 mL of Lysis buffer (50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 10% glycerol, 10 mM MgCl2) pre-chilled at 4 °C containing one EDTA-free protease inhibitor tablet.
- Lyse cells by sonication at 4 °C (time 7 min, 2 s on cycle, 2 s off cycle; amplitude: 70%).
- Clarify lysate by centrifugation at 26,900 x g for 45 min at 4 °C.
- Apply clarified lysate to a gravity flow column loaded with 10 mL of immobilized cobalt affinity resin, pre-equilibrated with Lysis buffer.
- Allow the supernatant to pass over the resin by gravity flow at 4 °C and wash the resin twice with 100 mL of Lysis buffer.
- Elute Nsa1 with 20 mL of Elution buffer (50 mM Tris-HCl pH 7.5, 500 mM NaCl, 5 mM MgCl2, 5% glycerol and 200 mM imidazole).
- Take 15 μL of the eluate and run it on a 4-15% SDS-PAGE gel to verify that the protein is eluted from the affinity resin (Figure 1A).
- Remove the MBP tag by TEV protease digestion. Add 1 mL of TEV protease19 (1 mg/mL stock) to the Nsa1 affinity resin elution and incubate it at 4 °C overnight.
- Concentrate MBP cleaved Nsa1 to ~ 5 mL using a centrifugal filter with a molecular weight cut off of 10 kDa.
- Apply MBP-cleaved Nsa1 to a gel-filtration column, pre-equilibrated in buffer A (20 mM Tris-HCl pH 7.5, 100 mM NaCl, 1 mM MgCl2, 5% glycerol and 1 mM β-mercaptoethanol) (Figure 1B).
- Analyze column fractions from gel filtration to verify that MBP is cleaved and separated from Nsa1 by running 15 μL samples on a 4-15% SDS-PAGE gel (Figure 1B).
- Combine fractions containing Nsa1 and concentrate to 8 mg/mL using a centrifugal filter with a molecular weight cut off of 10 kDa.
- Determine the protein concentration by measuring the absorbance at 280 nm on a spectrophotometer using the extinction coefficient 42530 M–1cm–1. Use protein immediately for proteolytic screening and crystallization trials.
2. Crystallization and Proteolytic Screening of Nsa 1
- Sparse Matrix Crystal Screening of Nsa1
- Centrifuge 500 μL of the 8 mg/mL stock of Nsa1 at 16,000 x g at 4 °C for 10 min.
- Setup crystallization trials using a crystallization robot and sparse matrix crystal screens. Fill the reservoir of 96 well trays with 30 μl of the individual crystal screen reagents from sparse matrix crystallization screens. Setup sitting drops with the robot by mixing 250 nL of the well solution with 250 nL of the protein solution.
- Seal the crystallization plates with tape and incubate them at 25 °C.
- Inspect the plates every two days for the first 2-3 weeks with a stereomicroscope.
- Verify that potential crystals hits contain protein with a UV microscope.
NOTE: For Nsa1 two different crystal forms (cubic and orthorhombic, Figure 2A) were obtained within 1 week from the following screens: JCSG+ condition B11 (1.6 M sodium citrate tribasic dehydrate, pH 6.5) and Wizard Precipitant Synergy Screen Block 2 condition C11 (20.1%(v/v) PEG 1500, 13.4%(v/v) PEG 400, 0.1 M Tris HEPES/sodium hydroxide, pH 7.5).
- Proteolytic Screening
NOTE: During crystallization optimization, it was discovered that the orthorhombic crystals of Nsa1 arose as a result of proteolytic cleavage and the crystals could not be duplicated using the full-length protein. Using a combination of limited proteolysis coupled with mass spectrometry, it was determined that the C-terminus of Nsa1 was sensitive to proteolysis and removal of the C-terminal tail was required for subsequent reproduction of the orthorhombic crystal form (Nsa1ΔC).- Prepare 1 mg/mL protease stock solutions of the following proteases α-chymotrypsin, trypsin, elastase, papain, subtilisin, and endoproteinase Glu-C.
- Create 1:10, 1:100, and 1:1000 dilutions of each 1 mg/mL protease stock with Dilution Buffer (10 mM HEPES, pH 7.5, 500 mM sodium chloride).
- Pipette 1 μL of protease stock (1:10, 1:100 and 1:1000) into 9 μL aliquots of protein (1 mg/mL) for each protease to be screened.
- Incubate the solution at 37 °C for 1 h.
- Stop the reaction by adding 10 μL of 2x SDS-PAGE sample buffer and heat the reaction at 95 °C for 5 min.
- Analyze the digests by running them on a 4-15% SDS-PAGE gel (Figure 2B).
- Identify protease resistant domains of the target protein by in-gel mass spectrometry analysis.
- Remove the protease-labile regions from the target protein by creating truncated expression constructs and following the cloning, expression, and purification protocol described above.
- Crystallization Optimization
- Prepare or obtain stock solutions of the following initial crystallization reagents: 1.6 M sodium citrate tribasic dehydrate pH 6.5, 100%(v/v) PEG 400, 50%(v/v) PEG 1500, 1 M HEPES/sodium hydroxide, pH 7.5.
- Prepare a stock solution of Nsa1FL and Nsa1ΔC at 8 mg/mL as described above.
- Optimize the Nsa1 cubic crystals (Nsa1FL).
- Prepare a 24-well grid screen with wells containing 500 µL with a gradient of 1-1.6 M of sodium citrate with pH 6.5 from a stock solution of 1.6 M sodium citrate tribasic with pH 6.5.
- Mix 1 µL of protein with 1 µL of well solution and place the mixture onto a siliconized cover slide. Carefully invert the cover slide on top of the pre-greased well and ensure it is well sealed but take care to not disturb the drop or break the cover slide. Repeat this process until the tray is filled and then store at 25 °C.
NOTE: Small cubic crystals should appear in 2-7 days. - Prepare a microseed stock of the cubic crystals. Use a mounted nylon loop to transfer ~10 small cubic crystals to a 1.5 mL micro-centrifuge tube containing a small bead and 50 µL of 1.6 M sodium citrate tribasic wit pH 6.5.
- Vortex the 1.5 mL micro-centrifuge tube at high speed (~ 3000 rpm) for 1 min.
- Make a series of 10-fold serial dilutions of the seed stock with 1.6 M sodium citrate tribasic and vortex the mixture thoroughly for 5 s.
- Fill each well of a 24-well grid, screen with 500 µL of 1.6 M sodium citrate tribasic pH 6.5.
- Optimize the microseeding conditions by setting up drops with varying ratios of protein with the seed stock solutions (Figure 3A). Larger cubic crystals of high diffraction quality should appear in 2-5 days (Figure 3C).
- Optimize the orthorhombic crystals (Nsa1ΔC)
- Prepare a grid screen with 500 µL in each well with 24 different conditions (Figure 3B) from stock solutions of 50%(v/v) PEG 1500, and 100%(v/v) PEG 400. In addition to gradients of PEG 1500 and PEG 400, each well should also contain 0.1 M HEPES/sodium hydroxide pH 7.5.
- Mix 1 µL of protein solution with 1 µL of well solution on a siliconized cover slide. Carefully invert the cover slide on top of the pre-greased well and ensure it is well sealed. Repeat this process until the entire tray has been filled. Store the trays at 25 °C.
NOTE: Nsa1 orthorhombic crystals should appear in 2-7 days. - Further optimize the orthorhombic crystals by microseeding as described for the cubic crystals (steps 2.3.3.3 to 2.3.3.7) using 500 µL of 20.1%(v/v) PEG 1500, 13.4%(v/v) PEG 400, and 0.1 M HEPES/sodium hydroxide pH 7.5 as the well solution.
NOTE: Nsa1 SeMet crystals should be optimized analogous to the native crystals.
3. X-ray Diffraction Data Collection and Nsa 1 Structure Solution
- Cryo-protection of Crystals and X-ray Diffraction Data Collection
- For the orthorhombic crystals (Nsa1ΔC), prepare a 1 mL cryoprotectant solution containing 22.5%(v/v) PEG 1500, 15%(v/v) PEG 400 and 0.1 M HEPES/sodium hydroxide pH 7.5.
- Fill a foam Dewar with liquid nitrogen, and pre-cool a crystal puck. Use caution when working with liquid nitrogen and wear protective gloves and goggles.
- Carefully invert the cover slide of the crystallization well containing crystals onto the stage of a stereomicroscope.
- Pipette 2 µL of the cryoprotectant solution onto a new cover slide.
- Attach a mounted nylon loop of the appropriate size for the crystal to a magnetic cryo wand.
- Using the aid of the stereomicroscope, quickly transfer a crystal to the cryoprotectant solution with the mounted cryo loop.
- Let the crystal equilibrate for 5 min in the cryoprotectant solution.
- Using the aid of the stereomicroscope, quickly loop the crystal from the cryoprotectant solution and plunge-freeze into liquid nitrogen.
- Wait for the liquid nitrogen around the loop to stop boiling and then release the loop from the wand into a specific location within the crystal puck.
- Cubic Nsa1FL crystals do not need to be cryoprotected and can be directly flash frozen (following steps 3.1.8-3.1.9 above).
- Seal the crystal puck using cryo tools and transfer to a shipping cane in a pre-chilled dewar. Store crystals in the pucks/dewars until data collection.
- If collecting data at a synchrotron, ship crystals to the synchrotron using a dry shipper.
- Collect X-ray diffraction data following standard techniques20.
NOTE: For Nsa1 native and SAD (single-wavelength anomalous dispersion) datasets were collected at 100 K on the SER-CAT beam lines 22-ID and 22-BM of the Advanced Photon Source at Argonne National Laboratory (Chicago, IL). The SAD Nsa1 dataset was collected at λ= 0.97911 Å. Data was recorded using a 1 s exposure time and 0.5° oscillations. The mosaicity of these crystals was typically around 0.3°. - Process and scale the X-ray diffraction images to generate reflection files for each data set in the appropriate space group.
NOTE: Nsa1 diffraction datasets were processed with HKL200021. The Nsa1 cubic crystals were processed in the space group P213 and the orthorhombic crystals were processed in the space group P212121.
- Nsa1 Structure Solution.
NOTE: There are several crystallography software packages that can be used to solve and refine crystal structures including Phenix and CCP422,23. Following is the protocol for Nsa1 structure solution using the Phenix software suite22.- Analyze the Native and SAD scaled datasets with phenix.xtriage22.
- Solve the structure of Nsa1 with Phenix.autosol using the SAD peak reflection file22,24. To run the program of AutoSol, input the number of selenomethionine sites (sites= 9), the fasta sequence file of Nsa1ΔC, and the wavelength used for SAD data collection (λ= 0.97911 Å).
NOTE: From the Nsa1 SAD dataset, phenix.autosol should be able to determine the experimental phases and build most of the model. - Make manual adjustments to the model with Coot25, followed by refinement in phenix.refine22,26.
- Solve the structure of the high-resolution native orthorhombic crystal and the cubic crystal by molecular replacement using Phaser27,28.
- After successful structure solution, inspect the model and the electron density map in Coot25.
- Build and refine the structures by running iterative rounds of refinement in phenix.refine22,26 and model building in Coot25.
4. SAXS Data Collection, Processing, and Modeling
- SAXS Data Collection
NOTE: SAXS data were recorded for full-length S. cerevisiae Nsa1 at the Advanced Light Source, on the high-throughput SIBYLS beamline 12.3.1 at Lawrence Berkeley National Laboratory, Berkeley, CA29.- Purify Nsa1FL following the protocol described above. 24 h prior to shipment of Nsa1FL to the beamline, run protein over a gel filtration column pre-equilibrated in buffer A.
- Pool fractions containing Nsa1 and determine the protein concentration as described earlier.
- Prepare 30 μl aliquots of a concentration series of Nsa1 from 1 to 6.2 mg/mL using buffer A.
- Transfer 20 μl of each concentration series of Nsa1 to a clear full skirt 96 well microplate along with buffer A alone controls.
NOTE: SAXS data is collected on dilute solutions using several concentrations of purified sample to avoid aggregation, inter-particle repulsion, and radiation damage effects. - Seal the microplate with a silicone sealing mat and ship overnight at 4 °C to the beamline.
- Store the microplate at 4 °C until data collection.
- Immediately before data collection, spin the plate at 3200 x g for 10 min at 4 °C to remove potential aggregates and air bubbles.
- Record SAXS data.
NOTE: For Nsa1, SAXS data were recorded for the buffer before and after each protein concentration series. Thirty-three consecutive scans of 0.3 s were collected for Nsa1FL over a concentration series (1 to 6.2 mg/mL) at 10 °C. - Perform buffer subtraction for SAXS data.
NOTE: For Nsa1 buffer subtraction was done automatically at the beamline but buffer subtraction can also be performed using data reduction software such as Scatter30 and the ATSAS suite31. Buffer subtraction is a critical part of SAXS data analysis and care must be taken to ensure that the buffer used for subtraction is identical to the protein sample buffer. - Average the 33 consecutive scans to create a *ave.dat file for each concentration.
NOTE: Overlay the 33 consecutive frames to compare the curves. Changes to the scattering curves over time is often indicative of radiation damage. Exclude these frames from averaging. Averaging of the Nsa1 consecutive scans was performed automatically at the SIBYLS beamline.
- SAXS Data Processing and Comparison of Concentration Series
NOTE: There are several software packages that can be used to analyze SAXS data. For Nsa1 the radius of gyration and pair-wise distance distribution functions were determined using PRIMUS32 and Gnom33 from the ATSAS 2.7.2 suite31 and compared across all protein concentrations to ensure there was no radiation damage or concentration-dependent inter-particle interactions10.- Open a terminal shell and go to the directory containing the SAXS data.
- Launch the PRIMUS32 GUI from within the terminal shell.
- Within the PRIMUS GUI, load the scattering curves (*ave.dat).
- Use AutoRg to determine the Radius of Gyration (Rg) and forward scattering intensity I(0), for each scattering curve (*ave.dat).
- Generate Kratky plots for each scattering curve (*ave.dat), to evaluate the degree of compactness.
- Use AutoGNOM33 to calculate the pair-wise distribution function (also called P(r)) for each scattering curve (*ave.dat). Enter a starting Dmax ≈ 3*Rg and optimize the Dmax value to obtain a smooth P(r) curve. During the optimization of Dmax, ensure that the calculated P(r) function is consistent with the experimental scattering curve by checking the reported χ2 value and visually assessing the overall fit to the scattering curve.
- Calculate the molecular weight of Nsa1 from the scattering curves using the volume of correlation34.
- Compare the structural parameters for each concentration to ensure that there are no radiation damage or concentration-dependent effects.
NOTE: Concentration effects can manifest as an increasing Rg and Dmax in relation to an increasing protein concentration. Forward scattering I(0) divided by the sample concentration should also remain constant across the protein concentration series.
- SAXS Modeling
NOTE: To determine the position of the C-terminus of Nsa1FL, the Nsa1 crystal structure10 (PDB 5SUM) and the SAXS scattering curves were used to carry out a combination of rigid body and ab initio modeling, using the programs BUNCH35 and Ensemble Optimization Method (EOM)36 from the ATSAS software suite31. The WD40 domain of Nsa1 was treated as a rigid body, while the C-terminus of Nsa1 along with several disordered loops from the WD40 domain were modeled to fit the experimental SAXS data.- Generate the input PDB file(s). The PDB file must be split every time residues are missing from the main chain, and the PDB file cannot contain multiple conformations, ligands, or water molecules.
- Run the program Pre_BUNCH35 to prepare the input PDB file for BUNCH (pre_bunch.pdb). Input the sequence of the target protein (*.seq), the number of domains/PDBs generated in 4.3.1, and each of the individual PDB files generated above.
- Calculate the scattering amplitudes for each individual PDB file using the program CRYSOL37. To run CRYSOL input the individual PDB file and the experimental SAXS scattering curve (*.dat). This will generate an amplitudes file (*.alm).
- Run the program BUNCH35 to model the WD40 domain of Nsa1 against the SAXS data using a combined rigid body and ab initio approach. Input the PDB file from Pre_BUNCH (pre_bunch.pdb), the experimental SAXS scattering curve (*.dat), and the individual amplitude (*.alm) files for each partial PDB file generated by CRYSOL.
- Compare the χ2 value from the starting PDB (5SUM) with that from the ab initio model generated by BUNCH using the experimental SAXS data.
NOTE: A successful BUNCH model should have a significantly lower χ2 value than the starting model. The theoretical scattering of the BUNCH model should also describe well the experimental data as judged by visual inspection of the overlay between the theoretical scattering of the BUNCH model to the SAXS data and its χ2 value (Figure 4, center pipeline). - Manually inspect the output files from BUNCH in Pymol38 to overlay the crystal structure with the model generated by BUNCH, and the SAXS envelope (Figure 4, center pipeline).
- Rerun BUNCH 10-20 times to generate independent models to confirm that the models are similar.
NOTE: For Nsa1 there was a range of χ2 values from ~1 to 3 from 20 independent runs. - Ensemble Modeling
NOTE: As an optional approach to BUNCH run either EOM36 or Minimal Ensemble Search39 (MES). EOM and MES use ensemble approaches, which are well suited for proteins with flexible domains/regions that are in multiple conformations.- Run the program EOM36 using the ATSAS online server. To run EOM, input the PDB files from 4.3.1, the sequence of the target protein (*.seq), and the experimental SAXS data (*.dat).
- Compare the χ2 values from the starting PDB (5SUM) with that from the ensemble generated by EOM using the experimental SAXS data.
NOTE: A successful EOM ensemble should have a significantly lower χ2 value than the starting model. The theoretical scattering of the ensemble should also describe well the experimental data as judged by visual inspection of the overlay between the theoretical scattering of the ensemble to the SAXS data and its χ2 value (Figure 4, right pipeline). One should also compare the χ2 values from BUNCH and EOM to determine which model best describes the experimental SAXS data. - Manually inspect the EOM conformers in Pymol38 to overlay the crystal structure with the conformers generated by EOM (Figure 4). Note the total number of conformers and the fraction of occupancy for each conformer that contributes to the scattering curve. For more rigid molecules, such as Nsa1, the number of conformers should be small (1-5) (Figure 4, right pipeline)36.
- Re-run EOM several times to ensure consistent results.
NOTE: For Nsa1 the number of conformers was typically 3 to 4 with χ2 values ranging from 0.1 to 0.3.
Representative Results
Nsa1 was PCR amplified from S. cerevisiae genomic DNA and subcloned into a vector containing an N-terminal 6x-Histidine affinity tag followed by MBP and a TEV protease site. Nsa1 was transformed into E. coli BL21(DE3) cells and high yields of protein expression were obtained following induction with IPTG and growth at 25 °C overnight (Figure 1A). Nsa1 was affinity-purified on immobilized cobalt affinity resin, followed by MBP cleavage with TEV protease, and finally resolved by size exclusion chromatography (Figure 1B). Fractions from size exclusion chromatography containing Nsa1 were pooled, concentrated to 8 mg/mL and then used for crystallization trials with a crystallization robot. Initial sparse matrix crystal screens yielded two different crystal forms of Nsa1, cubic and orthorhombic (Figure 2A).
During the optimization of the cubic and orthorhombic crystals, it was discovered that the orthorhombic crystals arose as the result of proteolytic cleavage of Nsa1. Limited proteolysis and mass spectrometry were used to determine the region of Nsa1 that was sensitive to proteolysis, and it was observed that Nsa1 was sensitive to a concentration gradient of the protease elastase (Figure 2B). Subsequent mass spectrometry analysis confirmed that this degradation resulted from loss of the C-terminus of Nsa1. A series of C-terminal truncations of Nsa1 were generated, to remove the proteolytic sensitive C-terminus (Figure 2C). The orthorhombic crystals could be repeated with the Nsa1ΔC (residues 1-434) truncation, which was ultimately used for SAD structure determination. The orthorhombic crystals could also be repeated by treating Nsa1FL with elastase for 1 hour at 4 °C prior to setting up crystal trays.
The cubic Nsa1 crystals were optimized using Nsa1FL through a combination of sodium citrate gradients, coupled with microseeding (Figure 3A). This yielded large, reproducible cubic crystals, with a diffraction limit of around 2.8 Å resolution (Figure 3C, left). The orthorhombic crystals could only be optimized using the C-terminal truncation variants of Nsa1, by varying the concentration gradients of PEG 1500 and PEG 400, combined with microseeding, which yielded large crystals with a diffraction limit of around 1.25 Å resolution (Figure 3A-C). Experimental phases of Nsa1 were determined by SeMet-SAD from a SeMet-derivative of Nsa1ΔC10.
The N-terminal seven-bladed β-propeller WD40 domain of Nsa1 was well resolved in both the cubic and orthorhombic crystal structures, however both structures lacked electron density for the C-terminus of Nsa1. SAXS was then used to determine the position of the missing C-terminal domain of Nsa1 in solution. After optimization of sample concentration for data collection, the partial atomic structure was used to perform rigid-body modeling, and generate an ab initio reconstruction of the missing components. The model was evaluated in terms of the goodness of the fit for the calculated scattering curves to the experimental data (Figure 4, center pipeline). The WD40 domain of Nsa1 alone is not a good fit of the experimental SAXS data, as evidenced by the discrepancy between the experimental scattering curve with the theoretical scattering curve, which was generated from crystal structure PDB ID 5SUM (Figure 4, left pipeline). In addition to rigid-body modeling, ensemble modeling was also done. This produced an ensemble of 3 to 4 conformers of Nsa1 and resulted in a lower χ2 value (Figure 4, right pipeline). The reduction in model discrepancy (χ2) using ensemble modeling revealed the conformational sampling of the Nsa1 C-terminal tail in solution.
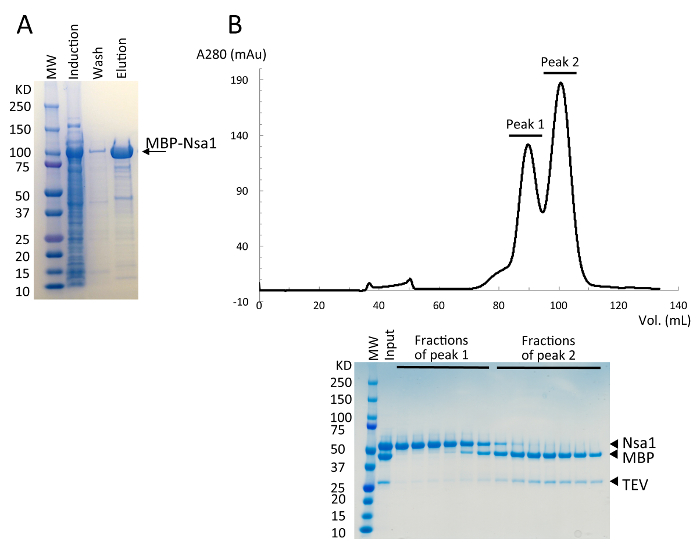
Figure 1. Expression and purification of Nsa1 with a 6X-His-MBP fusion tag. (A) SDS-PAGE analysis of the protein expression in BL21 (DE3) cells at 25 °C overnight and the first purification step using cobalt affinity resin. (B) Representative size exclusion chromatogram following TEV cleavage. The fractions from size exclusion chromatography were analyzed by SDS-PAGE. The fractions from peak 1 containing Nsa1 were collected and used for structural analysis. Please click here to view a larger version of this figure.
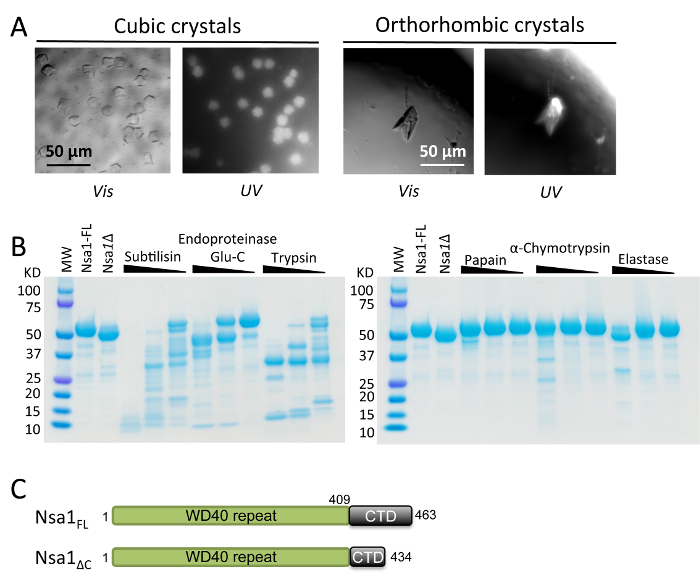
Figure 2. The C-terminus of Nsa1 is sensitive to proteolysis. (A) Initial crystallization trials of Nsa1 yielded two different crystals forms: cubic and orthorhombic. UV microscope was used to verify that the crystals contained protein. Vis: Visible light, UV: UV microscopes. Scale bar = 50 µm in each window. (B) Proteolytic screening analyzed by SDS-PAGE. Three dilutions (1:10, 1:100, 1:1000) for each protease stock (0.1, 0.01, 0.001 mg/mL) were combined with aliquots of protein (1 mg/mL) to be screened. Protease resistant domains were analyzed the by SDS-PAGE and mass spectrometry after 37 °C incubation for 60 min. Nsa1-FL: fresh purified protein, Nsa1Δ: purified protein stored at 4 °C for 3 weeks after which a degraded form of the protein was observed. (C) Schematic diagram of the Nsa1 full-length (upper) and the C-terminal truncation construct (lower). Please click here to view a larger version of this figure.
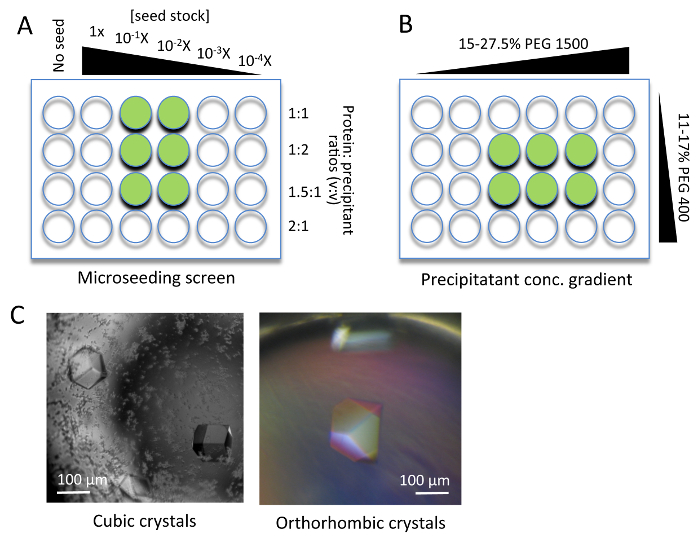
Figure 3. Nsa1 Crystallization Optimization. (A) A seed stock was prepared from initial small crystals and used to make a dilution series (1x ~ 1/104x) (microseeding). By mixing 1 µL of protein with 1 µL of the diluted seed stock, the bigger single crystals grew within a week. (B) Precipitant concentration gradient for orthorhombic crystal optimization. (C) Optimized cubic and orthorhombic crystals used for data collection. Green circles indicate the typical area of the crystal trays which yielded data collection quality crystals. Please click here to view a larger version of this figure.
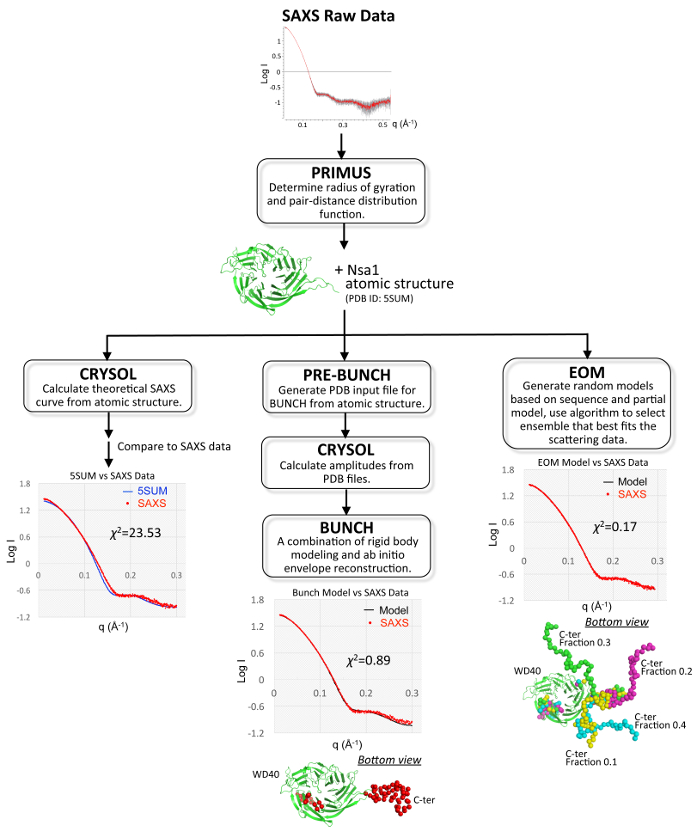
Figure 4. Schematic of Nsa1 SAXS Analysis. Overview of the pipeline used to process SAXS data and generate models with BUNCH (center) and EOM (right). The left pipeline shows the discrepancy between the experimental scattering curve (red circles, protein concentration: 6 mg/mL) with the theoretical scattering curve (blue line), which was generated from the crystal structure PDB ID 5SUM. The models were evaluated by comparing the experimental SAXS scattering curve (red circles, protein concentration: 6 mg/mL) with the scattering curve derived from the BUNCH model of Nsa1 (black line) or the EOM conformers of Nsa1 (black line). In each model, the WD40 domain is shown in cartoon colored in green (PDB ID 5SUM), the flexible C-terminus is shown in spheres colored in red for BUNCH and green, magenta, cyan, and yellow for the individual EOM conformers. The fraction of each conformer derived from EOM is labeled next to the model. Please click here to view a larger version of this figure.
Discussion
Using this protocol, recombinant Nsa1 from S. cerevisiae was generated for structural studies by both X-ray crystallography and SAXS. Nsa1 was well-behaved in solution and crystallized in multiple crystal forms. During the optimization of these crystals, it was discovered that the C-terminus of Nsa1 was sensitive to protease degradation. The high resolution, orthorhombic crystal form could only be duplicated with C-terminal truncation variants of Nsa1, likely because the flexible C-terminus of Nsa1 prevented crystal packing. The structure of Nsa1 was solved by X-ray crystallography to high resolution, but the C-terminus could not be built in either crystal form because it was not ordered. Crystallography is the premiere technique for determining atomic resolution structures of macromolecules around the size of Nsa1, however as with any method, crystallography does have some limitations. One of the major limitations of crystallography is the inability to resolve disordered regions of proteins40,41.
The C-terminus of Nsa1 is important for proper nucleolar localization of the protein, underscoring the need to study its structure10. The C-terminus of Nsa1, was resolved by SAXS, a complementary structural biology technique to X-ray crystallography. SAXS data was recorded for full-length Nsa1 across a concentration series. From this concentration series, the optimal concentration for Nsa1 SAXS data collection and processing was determined. SAXS data were recorded for Nsa1 at 6, 4.5, and 3.0 mg/mL. The Guinier region, P(r) function and molecular weight were determined across the concentration series to ensure that the sample was well-behaved and not aggregated under the experimental conditions tested. To reconstruct the full-length structure of Nsa1, the theoretical scattering amplitude was determined from the partial crystal structure and then ab initio methods were used to model the flexible C-terminus. From this hybrid approach, it was determined that the flexible C-terminus of Nsa1 extends outward from the ordered WD40 domain.
Advances in processing tools has driven the popularity of SAXS for macromolecular structural studies. SAXS measures the X-ray scattering pattern from randomly oriented protein in solution to provide low-resolution structural information, including molecular mass and overall shape. Consequently, SAXS has emerged as a powerful orthogonal structure validation tool for crystallography. This is largely due to the development of computational methods to calculate the theoretical scattering of atomic structures and comparing them to experimental SAXS data37. Using this approach, the conformational state, quaternary structure, and higher-order assembly observed in a crystal lattice can be compared to the structural characteristics of the particle in solution. Furthermore, disordered loops and termini missing in high-resolution structures determined by X-ray crystallography can be modeled using solution scattering data. This hybrid structural approach uses the crystal structure as a building block for SAXS-guided modeling of missing residues and has proven to be effective in mapping the C-terminus of Nsa110, as well as other macromolecules such as the influenza A virus M1 matrix protein42and DEAD-box RNA chaperones43. Advanced SAXS-based modeling software can also address more complex systems, such as intrinsically disordered proteins, by mapping the conformational landscape of these systems using a series of conformers that together contribute to the overall scattering potential of the particle in solution16,39. Taken together, recent advances in SAXS data collection and processing tools contributes to the success of hybrid structural biology approaches for tackling challenging biological systems.
The combination of solution scattering with high resolution structures is poised to answer important questions about the flexibility and dynamics of macromolecules44. Many proteins, such as Nsa1, have dynamic regions that are important for biological function. In this manuscript a template protocol is provided which details the combination of SAXS with high-resolution structure determination by X-ray crystallography. In addition to X-ray crystallography SAXS can also be used to compliment other structural biology techniques including NMR, electron paramagnetic resonance (EPR), and fluorescence resonance energy transfer (FRET), further highlighting the importance of SAXS as a complementary structural biology technique45,46,47.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Diffraction data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID and 22-BM beamlines at the Advanced Photon Source (APS), Argonne National Laboratory. The SAXS data was collected on the SIBYLS beamline at the Advance Light Source (ALS), Lawrence Berkeley National Laboratory. We would like to thank the staff at the SIBYLS beamline for their help with remote data collection and processing. We are grateful to the National Institute of Environmental Health Sciences (NIEHS) Mass Spectrometry Research and Support Group for help determining the protein domain boundaries. This work was supported by the US National Institute of Health Intramural Research Program; US National Institute of Environmental Health Sciences (NIEHS) (ZIA ES103247 to R. E. S.) and the Canadian Institutes of Health Research (CIHR, 146626 to M.C.P). Use of the APS was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. W-31-109-Eng-38. Use of the Advanced Light Source (ALS) was supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Additional support for the SIBYLS SAXS beamline comes from the National Institute of Health project MINOS (R01GM105404) and a High-End Instrumentation Grant S10OD018483. We would also like to thank Andrea Moon and Dr. Sara Andres for their critical reading of this manuscript.
Materials
| Molecular Cloning of Nsa1 | |||
| pMBP2 parallel vector | Sheffield et al, Protein Expression and Purification 15, 34-39 (1999) | We used a modified version of pMBP2 which included an N-terminal His-tag (pHMBP) | |
| S. cerevisiae genomic DNA | ATCC | 204508D-5 | |
| Primers for cloning Nsa1 | |||
| SC_Nsa1_FLFw | IDT | CGC CAA AGG CCT ATGAGGTTACTAGTCAGCTGTGT GGATAG |
|
| SC_Nsa1_FLRv | IDT | AATGCAGCGGCCGCTCAAATTTT GCTTTTCTTACTGGCTTTAGAAGC AGC |
|
| SC_Nsa1_DeltaCFw | IDT | GGGCGCCATGGGATCCATGAGG TTACTAGTCAGCTGTGTGG |
|
| SC_Nsa1_DeltaCRv | IDT | GATTCGAAAGCGGCCGCTTAAAC CTTCCTTTTTTGCTTCCC |
|
| Recombinant Protein Production and Purification of Nsa1 | |||
| Escherichia coli BL21 (DE3) Star Cells | Invitrogen | C601003 | |
| pMBP- NSA1 and various truncations | Lo et al., 2017 | ||
| Selenomethionine | Molecular Dimensions | MD12-503B | |
| IPTG, Dioxane-Free | Promega | V3953 | |
| EDTA Free Protease Inhibitor Cocktail | Sigma-Aldrich | 4693159001 | |
| Sodium Chloride | Caledon Laboratory Chemicals | 7560-1-80 | |
| Magnesium Chloride hexahydrate | Sigma-Aldrich | M2670 | |
| Tris Buffer, 1 M pH7.5 | KD Medical | RGF-3340 | |
| Glycerol | Invitrogen | 15514-029 | |
| beta-mercaptoethanol | Sigma | M6250 | |
| 1M Imidazole, pH 8.0 | Teknova | I6980-06 | |
| Talon Affinity Resin | Clonetech | 635503 | |
| Amicon Ultra 15 mL Centrifugal Filter (MWCO 10K) | Millipore | UFC901024 | |
| HiLoad 16/600 Superdex 200 Prep Grade Gel Filtration Column | GE-Healthcare | 28989335 | |
| TEV Protease | Prepared by NIEHS Protein Expression Core | Expression plasmid provided by NCI (Tropea et al. Methods Mol Biology, 2009) | |
| 4-15% Mini-PROTEAN TGX Precast Protein Gels | BioRad | 456-8056 | |
| Crystallization, Proteolytic Screening | |||
| Crystal Screen | Hampton Research | HR2-110 | |
| Crystal Screen 2 | Hampton Research | HR2-112 | |
| Salt Rx | Hampton Research | HR2-136 | |
| Index Screen | Hampton Research | HR2-144 | |
| PEG/Ion Screen | Hampton Research | HR2-139 | |
| JCSG+ | Molecular Dimensions | MD1-37 | |
| Wizard Precipitant Synergy | Molecular Dimensions | MD15-PS-T | |
| Swissci 96-well 3-drop UVP sitting drop plates | TTP Labtech | 4150-05823 | |
| 3inch Wide Crystal Clear Sealing Tape | Hampton Research | HR4-506 | |
| Proti-Ace Kit | Hampton Research | HR2-429 | |
| PEG 1500 | Molecular Dimensions | MD2-100-6 | |
| PEG 400 | Molecular Dimensions | MD2-100-3 | |
| HEPES/sodium hydroxide pH 7.5 | Molecular Dimensions | MD2-011- | |
| Sodium Citrate tribasic | Molecular Dimensions | MD2-100-127 | |
| 22 mm x 0.22 mm Siliconized Coverslides | Hampton Research | HR3-231 | |
| 24 Well Plates with sealant (VDX Plate with Sealant) | Hampton Research | HR3-172 | |
| 18 mM Mounted Nylon Loops (0.05 mm to 0.5 mM) | Hampton Research | HR4-945, HR4-947, HR4-970, HR4-971 | |
| Seed Bead Kit | Hampton Research | HR2-320 | |
| Magnetic Crystal Caps | Hampton Research | HR4-779 | |
| Magnetic Cryo Wand | Hampton Research | HR4-729 | |
| Cryogenic Foam Dewar | Hampton Research | HR4-673 | |
| Crystal Puck System | MiTeGen | M-CP-111-021 | |
| Full Skirt 96 well Clear Plate | VWR | 10011-228 | |
| AxyMat Sealing Mat | VWR | 10011-130 | |
| Equipment | |||
| UVEX-m | JAN Scientific, Inc. | ||
| Nanodrop Lite Spectrophotometer | Thermo-Fisher | ||
| Mosquito Robot | TTP Labtech | ||
| Software/Websites | |||
| HKL2000 | Otwinoski and Minor, 1997 | ||
| Phenix | Adams et al., 2010 | ||
| Coot | Emsley et al., 2010 | ||
| ATSAS | Petoukhov et al., 2012 | https://www.embl-hamburg.de/biosaxs/atsas-online/ | |
| Scatter | Rambo and Tainer, 2013 | ||
| Pymol | The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC. | ||
| BUNCH | Petoukhov and Svergun, 2005 | ||
| CRYSOL | Svergun et al, 1995 | ||
| PRIMUS | Konarev et al, 2003 | ||
| EOM | Tria et al, 2015 |
References
- Thomson, E., Ferreira-Cerca, S., Hurt, E. Eukaryotic ribosome biogenesis at a glance. J Cell Sci. 126 (Pt 21), 4815-4821 (2013).
- Woolford, J. L., Baserga, S. J. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. 遗传学. 195 (3), 643-681 (2013).
- Kressler, D., Hurt, E., Bassler, J. A Puzzle of Life: Crafting Ribosomal Subunits. Trends Biochem Sci. , (2017).
- Tomecki, R., Sikorski, P. J., Zakrzewska-Placzek, M. Comparison of preribosomal RNA processing pathways in yeast, plant and human cells – focus on coordinated action of endo- and exoribonucleases. FEBS Lett. , (2017).
- Kressler, D., Hurt, E., Bassler, J. Driving ribosome assembly. Biochim Biophys Acta. 1803 (6), 673-683 (2010).
- Kressler, D., Roser, D., Pertschy, B., Hurt, E. The AAA ATPase Rix7 powers progression of ribosome biogenesis by stripping Nsa1 from pre-60S particles. J Cell Biol. 181 (6), 935-944 (2008).
- Hiraishi, N., Ishida, Y., Nagahama, M. AAA-ATPase NVL2 acts on MTR4-exosome complex to dissociate the nucleolar protein WDR74. Biochem Biophy Res Co. 467 (3), 534-540 (2015).
- Maserati, M., et al. Wdr74 is required for blastocyst formation in the mouse. PLoS One. 6 (7), e22516 (2011).
- Weinhold, N., Jacobsen, A., Schultz, N., Sander, C., Lee, W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat Genet. 46 (11), 1160-1165 (2014).
- Lo, Y. H., Romes, E. M., Pillon, M. C., Sobhany, M., Stanley, R. E. Structural Analysis Reveals Features of Ribosome Assembly Factor Nsa1/WDR74 Important for Localization and Interaction with Rix7/NVL2. Structure. 25 (5), 762-772 (2017).
- Lander, G. C., Saibil, H. R., Nogales, E. Go hybrid: EM, crystallography, and beyond. Curr Opin Struc Biol. 22 (5), 627-635 (2012).
- Putnam, C. D., Hammel, M., Hura, G. L., Tainer, J. A. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 40 (3), 191-285 (2007).
- Jaskolski, M., Dauter, Z., Wlodawer, A. A brief history of macromolecular crystallography, illustrated by a family tree and its Nobel fruits. FEBS J. 281 (18), 3985-4009 (2014).
- Zheng, H., et al. X-ray crystallography over the past decade for novel drug discovery – where are we heading next?. Expert Opin Drug Dis. 10 (9), 975-989 (2015).
- Kikhney, A. G., Svergun, D. I. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 589 (19 Pt A), 2570-2577 (2015).
- Bernado, P., Mylonas, E., Petoukhov, M. V., Blackledge, M., Svergun, D. I. Structural characterization of flexible proteins using small-angle X-ray scattering. J Am Chem Soc. 129 (17), 5656-5664 (2007).
- Sheffield, P., Garrard, S., Derewenda, Z. Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expres Purif. 15 (1), 34-39 (1999).
- Doublie, S. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 276, 523-530 (1997).
- Tropea, J. E., Cherry, S., Waugh, D. S. Expression and purification of soluble His(6)-tagged TEV protease. Methods Mol Biol. 498, 297-307 (2009).
- Wlodawer, A., Minor, W., Dauter, Z., Jaskolski, M. Protein crystallography for aspiring crystallographers or how to avoid pitfalls and traps in macromolecular structure determination. FEBS J. 280 (22), 5705-5736 (2013).
- Otwinowski, Z., Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Macromolecular Crystallography, Pt A. 276, 307-326 (1997).
- Adams, P. D., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. 66 (Pt 2), 213-221 (2010).
- Winn, M. D., et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D. 67 (Pt 4), 235-242 (2011).
- Terwilliger, T. C., et al. Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallographica Section D. 65, 582-601 (2009).
- Emsley, P., Lohkamp, B., Scott, W. G., Cowtan, K. Features and development of Coot. Acta Crystallogr D. 66 (Pt 4), 486-501 (2010).
- Afonine, P. V., et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D. 68 (Pt 4), 352-367 (2012).
- McCoy, A. J. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D. 63 (Pt 1), 32-41 (2007).
- McCoy, A. J., et al. Phaser crystallographic software. J Appl Crystallogr. 40 (Pt 4), 658-674 (2007).
- Dyer, K. N., et al. High-throughput SAXS for the characterization of biomolecules in solution: a practical approach. Methods Mol Biol. 1091, 245-258 (2014).
- Forster, S., Apostol, L., Bras, W. Scatter: software for the analysis of nano- and mesoscale small-angle scattering. J Appl Crystallogr. 43, 639-646 (2010).
- Petoukhov, M. V., et al. New developments in the ATSAS program package for small-angle scattering data analysis. J Appl Crystallogr. 45, 342-350 (2012).
- Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J., Svergun, D. I. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr. 36, 1277-1282 (2003).
- Svergun, D. I. Determination of the Regularization Parameter in Indirect-Transform Methods Using Perceptual Criteria. J Appl Crystallogr. 25, 495-503 (1992).
- Rambo, R. P., Tainer, J. A. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 496 (7446), 477-481 (2013).
- Petoukhov, M. V., Svergun, D. I. Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys J. 89 (2), 1237-1250 (2005).
- Tria, G., Mertens, H. D., Kachala, M., Svergun, D. I. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ. 2 (Pt 2), 207-217 (2015).
- Svergun, D., Barberato, C., Koch, M. H. J. CRYSOL – A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr. 28, 768-773 (1995).
- . The PyMOL Molecular Graphics System Version 1.8 Available from: https://pymol.org (2015)
- Pelikan, M., Hura, G. L., Hammel, M. Structure and flexibility within proteins as identified through small angle X-ray scattering. Gen Physiol Biophys. 28 (2), 174-189 (2009).
- Deller, M. C., Kong, L., Rupp, B. Protein stability: a crystallographer’s perspective. Acta Crystallogr F. 72 (Pt 2), 72-95 (2016).
- Hinsen, K. Structural flexibility in proteins: impact of the crystal environment. Bioinformatics. 24 (4), 521-528 (2008).
- Shtykova, E. V., et al. Structural analysis of influenza A virus matrix protein M1 and its self-assemblies at low pH. PLoS One. 8 (12), e82431 (2013).
- Mallam, A. L., et al. Solution structures of DEAD-box RNA chaperones reveal conformational changes and nucleic acid tethering by a basic tail. Proc Natl Acad Sci U S A. 108 (30), 12254-12259 (2011).
- Papaleo, E., et al. The Role of Protein Loops and Linkers in Conformational Dynamics and Allostery. Chem Rev. 116 (11), 6391-6423 (2016).
- Rozycki, B., Boura, E. Large, dynamic, multi-protein complexes: a challenge for structural biology. J Phys Condens Matter. 26 (46), 463103 (2014).
- Schlundt, A., Tants, J. N., Sattler, M. Integrated structural biology to unravel molecular mechanisms of protein-RNA recognition. Methods. 118, 119-136 (2017).
- Thompson, M. K., Ehlinger, A. C., Chazin, W. J. Analysis of Functional Dynamics of Modular Multidomain Proteins by SAXS and NMR. Methods Enzymol. 592, 49-76 (2017).
