A cristalografia de macromoléculas à temperatura ambiente (TR) é novamente popular na comunidade de biologia estrutural. O desenvolvimento de fontes de luz XFEL (X-ray Free Electron Laser) estimulou o desenvolvimento de abordagens de entrega de amostras de RT 1,2,3,4, e esses métodos têm sido aplicados a síncrotrons 5,6,7,8. Não só os métodos de RT abrem a possibilidade de estrangulamentos experimentais bomba-sonda 9,10,11,12, mas também há evidências crescentes de que eles promovem estados conformacionais alternativos dentro das proteínas 13,14,15,16,17.
No entanto, a principal razão pela qual os criométodos ganharam força sobre as abordagens de RT no final da década de 1990 foi a desaceleração dos danos da radiação por temperaturas cristalinas abaixo de zero18. O criométodo19 começou a permitir a coleta de um conjunto completo de dados a partir de um único cristal de proteína. Métodos modernos de RT em XFELs e síncrotrons resolveram o problema dos danos por radiação monocristalina através do desenvolvimento de estratégias rápidas de liberação de cristais (> 100 Hz) 1,2,3,4. Esses métodos permitem a coleta de um conjunto de dados completo a partir de milhares de cristais expostos individualmente. Essas abordagens de entrega de RT requerem, portanto, a produção de grandes quantidades de soluções contendo microcristais homogêneos (> 100 μL de < cristais de 50 μm). No entanto, como os criométodos tendem a exigir apenas cristais individuais, os métodos para criar tais polpas microcristalinas não são atualmente onipresentes nos laboratórios de cristalografia de proteínas.
Existem exemplos na literatura de como realizar partes do procedimento de otimização da microcristalização para amostras seriadas de cristalografia. Aqui, deve-se fazer uma distinção entre proteínas de membrana e proteínas solúveis. Protocolos para otimizar o crescimento de cristais de proteína de micromembrana cultivados em monooleína (ou algum outro lipídio), para fase cúbica lipídica (LCP), têm sido bem descritos20,21,22. No entanto, métodos para a microcristalização de proteínas solúveis, incluindo proteínas de membrana cultivadas em condições não-LCP, são geralmente escassos. Estudos anteriores focaram em partes específicas do processo, como a triagem de microcristais 23,24, o aumento da nucleação 24 e a escala usando difusão em interface livre 25, mas não um método completo.
No entanto, recentemente foi descritoum método 26 que tenta oferecer um protocolo completo. Como muitos aspectos da cristalografia de proteínas, ela não é nova. Muitas das ideias propostas já foram descritas por Rayment (2002)27. O método visa mostrar aos cristalógrafos como realizar a conversão de um único cristal cultivado usando difusão de vapor, para uma metodologia em batelada para cultivar milhares de microcristais. O método enfoca a difusão de vapor como um ponto de partida comum, já que 95% de todas as deposições do Protein Data Bank (PDB) provêm de cristais crescidos em placas de difusão de vapor26. A difusão de vapor não é, no entanto, o método ideal para microcristalização26, por isso é descrita uma metodologia para converter a difusão de vapor em cristalização em batelada. Uma vez que os cristais podem ser cultivados em lote, as rotas de escala para volumes maiores tornam-se mais viáveis. Dado os caprichos da cristalização de proteínas, os autores enfatizam que este método não é à prova de falhas. No entanto, o protocolo deve, pelo menos, fornecer uma visão sobre o “espaço de cristalização” de uma proteína.
Este método baseia-se no diagrama de fase de cristalização de proteínas e em como a compreensão desse diagrama pode atuar como um guia durante a otimização da microcristalização. Um diagrama de fase de proteína é comumente representado como um gráfico x/y com concentrações precipitantes e proteicas nos eixos x e y, respectivamente (Figura 1A). A partir do ponto de água pura (canto inferior esquerdo – Figura 1A), a concentração de proteína e precipitante aumenta até atingir a linha de solubilidade. A linha de solubilidade marca o ponto de supersaturação (linha roxa – Figura 1A). Quando uma proteína é supersaturada, a solução torna-se termodinamicamente instável e começará a separar-se em duas fases: “rica em proteínas” e uma solução saturada estável. Essa separação pode ocorrer em qualquer lugar além da linha de solubilidade e sua cinética depende das propriedades da proteína e dos componentes da solução.
Quando as concentrações de proteína e precipitante são muito grandes, a proteína se decompõe de forma instável fora da solução e resulta em precipitado amorfo (região rósea – Figura 1A). No entanto, a separação ordenada de fases pode ocorrer na região de nucleação [ver Garcia-Ruiz (2003)28 para descrição detalhada] e os nucleantes cristalinos têm propensão a se formar (região verde – Figura 1A). A nucleação e o crescimento removem a proteína da solução e movem a gota para a região metaestável, onde o crescimento pode continuar até que a linha de solubilidade seja atingida [ver McPherson e Kuznetsov (2014)29 para discussão detalhada]. O diagrama é, para a grande maioria das condições de cristalização, uma simplificação grosseira30. Apesar disso, no entanto, o diagrama ainda é de grande utilidade para os microcristalógrafos, pois o mapeamento do diagrama permite determinar a linha de solubilidade e a cinética de nucleação.
Em termos de criação de microcristais, os dois fatores durante a cristalização que precisam ser otimizados são o número de cristais (Xn) e sua dimensão média e mais longa (Xs). X n será proporcional ao número de eventos de nucleação (n ) (Eq. 1).
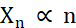 Eq. 1
Eq. 1
X s é proporcional à concentração de proteína livre acima da linha de solubilidade (Ps) dividida por Xn (Eq. 2).
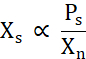 Eq. 2
Eq. 2
Em uma situação perfeita, cada evento de nucleação produziria um cristal possível e cada um desses cristais, teria igual acesso à proteína disponível em solução. A Figura 2 é uma representação gráfica de um cenário ideal da relação entre Xn e Xs. Na prática, o principal controle que um cristalógrafo tem sobre Xn e Xs é influenciando a quantidade de nucleação ou pela adição de cristais de sementes. O microcristalógrafo deve julgar como aumentar Xn de tal forma que uma concentração de cristal adequada e tamanho de cristal possam ser criados.
A maioria das técnicas de cristalização requer um “período de transição” (Figura 1B). Por exemplo, em um experimento de difusão de vapor, ao misturar a proteína e as soluções precipitantes, as concentrações de cada uma mudam à medida que a gota se equilibra com a solução do poço. Espera-se que essas mudanças façam a transição gradual da queda para a zona de nucleação, onde a propensão para cristalização aumentará. À medida que os cristais começam a se nuclear e crescem, a quantidade de proteína em solução começará a cair, diminuindo a probabilidade de nucleação adicional. A quantidade final de nucleação será específica da proteína e da condição, e também dependente da profundidade de penetração na zona de nucleação. Dada a penetração limitada da zona de nucleação de métodos que requerem uma etapa de transição, o nível de nucleação será limitado à taxa de nucleação no limite da região de nucleação metaestável.
Devido à importância de ser capaz de aumentar o nível de nucleação para um microcristalógrafo, é importante passar para uma metodologia de cristalização em batelada. O lote pode aproveitar melhor toda a região de nucleação (Figura 1C). Em métodos em batelada, a ideia é misturar a proteína e o precipitante de forma que uma solução supersaturada seja criada sem a necessidade de mudanças nas concentrações dos componentes. A nucleação deve ser possível imediatamente após a mistura. Os métodos em lote permitem, portanto, que toda a zona de nucleação seja teoricamente atingida. Qualquer aumento na cinética de nucleação além do limite de nucleação metaestável pode então ser utilizado.
Se o nível basal de nucleação de cristais não for suficiente para gerar um Xn grande, métodos de micro-semeadura podem ser usados. Na micro-semeadura, cristais pré-cultivados são quebrados para criar uma lama de fragmentos cristalinos que podem atuar como arcabouço para o crescimento de cristais frescos31,32. A microsemeadura tem sido amplamente utilizada na preparação seriada de amostras cristalográficas como forma de aumentar Xn sem a necessidade de aumentar a nucleação de cristais (Figura 1C).
A transição da difusão de vapor para a batelada pode ser visualizada em um diagrama de fases como movendo o ponto de partida experimental das regiões não supersaturadas ou metaestáveis para a zona de nucleação. Isso pode ser feito aumentando-se as concentrações de proteínas e/ou precipitantes e/ou a relação dos dois dentro da gota (Figura 1D), observando-se em quais condições os cristais aparecem rapidamente (< 24 h)26. O equilíbrio completo da gota de difusão do vapor pode levar dias ou semanas33. Portanto, ao procurar condições que mostrem cristais aparecendo rapidamente, condições de lote podem ser encontradas sem ter que passar para formatos alternativos de triagem de cristalização, como micro-lote34,35,36,37.
Uma vez que a zona de nucleação foi encontrada, uma condição de lote foi encontrada e um morfograma – aqui, um diagrama de fase aproximado – pode ser criado. O morfograma é de grande utilidade quando se pensa em utilizar um protocolo de batelada semeada ou batelada reta. Plotando o Xn em função da proteína e da concentração precipitante, pode-se avaliar a cinética de nucleação26. Se X n permanecer baixo em toda a região de nucleação, o lote semeado pode ser necessário para tornar Xn grande o suficiente para limitar o crescimento de cristais. Essa avaliação é o primeiro passo no processo de escalonamento para volumes maiores (> 100 μL).
Este método foi projetado de tal forma que pudesse ser conduzido na maioria dos laboratórios de cristalização usando equipamento padrão de cristalização por difusão de vapor. Muitos estudos também têm sido realizados descrevendo técnicas para facilitar muitas partes desse processo, caso o equipamento esteja disponível. Estes incluem, mas não estão limitados a, espalhamento dinâmico de luz (DLS)25,27, imagem não-linear 20,24,25, difração de pó 20,24,27 e microscopia eletrônica 26 [ver Cheng et al. (2020)40 para uma bela revisão].
O objetivo deste trabalho é fornecer uma demonstração visual do método de transição da cristalização por difusão de vapor de pequeno volume ( de 100 μL). A endotiapepsina de Cryphonectria parasitica tem sido usada como um sistema de exemplo para demonstrar essa tradução. O tipo de experimento e o método de entrega da amostra para os quais os microcristais são necessários influenciarão a saída idealde X s 26. Para experimentos de mistura que requerem uma resolução de tempo de milissegundos41 ou bicos virtuaisdinâmicos a gás42, um X s final de < 5 μm pode ser desejável. Neste caso, o objetivo era produzir cristais de proteína que difratassem a aproximadamente 1,5 Å, para um experimento de bomba-sonda ativada por fótons, e usando uma abordagem de entrega de alvo fixo.
Para ilustrar as necessidades amostrais de um experimento de cristalografia seriado usando endotiapsina, a Tabela 1 mostra os parâmetros experimentais de um experimento hipotético. As informações da amostra foram baseadas no protocolo descrito a seguir. Dadas algumas estimativas conservadoras sobre as taxas de acerto e os requisitos de coleta de dados, 50 mg é a estimativa do consumo total da amostra para todo o experimento.
A Figura 3 mostra um fluxograma do processo completo de otimização, desde a cristalização inicial por difusão de vapor de pequeno volume até o lote em grande escala. Para a maioria dos projetos de cristalografia seriada, este protocolo começará na Etapa 2: ‘transição para lote’, uma vez que a proteína alvo já terá sido cristalizada. No entanto, o Passo 1 foi incluído para completude e para lembrar os leitores de sua importância. Encontrar uma condição que dê origem a um cristal bem difratante, único e grande é o melhor ponto de partida para a otimização de microcristais. Na Etapa 2, essa condição pode então ser otimizada da difusão de vapor para batelada, e um morfograma das regiões de nucleação e metaestável pode ser plotado. Feito isso, o dimensionamento da condição de lote para volumes maiores pode ser executado na Etapa 3. Ao final do fluxograma, um cristalógrafo terá criado um protocolo de microcristalização repetível, de grande volume (> de 100 μL), de microcristalização, em lote para endotiapsina. Este método pode então ser aplicado à sua proteína particular de interesse.
