長年にわたり、シングルセル技術は生物学的プロセスの分析のゴールドスタンダードでした。当初は、顕微鏡検査、フローサイトメトリー、および同様のアッセイによる単一細胞表現型に限定されていました。シングルセル解析におけるブレークスルーは、シングルセル分子プロファイリングのアプローチ、特に個々の細胞のトランスクリプトーム全体の特性評価を可能にするシングルセルRNAシーケンシング(scRNA-Seq)の開発によってもたらされました。非常に強力なscRNA-Seqは、特定の条件と時点における細胞の転写状態に関する情報を生成します。しかし、転写を駆動する遺伝子制御や、時間の経過とともに起こる分子修飾については可視化できません。この制限を克服するために、同じ細胞から複数の因子やプロセスを解析できるシングルセルマルチオミクスアッセイの開発に多くの努力が払われてきました1,2,3,4。単一細胞内の2つのモダリティの測定に初めて成功したのは、CITE-Seqアプローチ5において、マルチプレックス表面タンパク質発現パターンを個々の細胞の完全なトランスクリプトームと結びつけることでした。最近の進化では、遺伝子発現とクロマチンアクセシビリティが組み合わされ(シーケンシングを用いたトランスポザーゼアクセス可能クロマチンアッセイ、ATAC-Seq)、それによって同じ細胞内のトランスクリプトームモダリティとエピゲノムモダリティを同時に捕捉するようになりました(例:sci-CAR)6。トランスクリプトミクスを細胞の表現型または同じ細胞のエピジェネティックな変化と関連付けることを可能にした最初の商用ソリューションは、10X Genomicsから生まれました。
単一細胞分子プロファイリングのための実験には、以下のステップが含まれます:(1)組織の解離または単一細胞懸濁液の調製;(2)細胞精製および/または核単離。(3)パーティショニングとバーコード化。(4)図書館の建設と品質管理。(5)次世代シーケンシング;(6)データ分析。ステップ(3)〜(6)は、採用する技術によって大きく異なる場合がありますが、最初のステップは一般的にそれらすべてに共通です。調製された細胞/核懸濁液の品質は、実験の全体的な結果を決定します。組織の種類によっては、高品質の単一細胞/核懸濁液を得ることが困難な場合があります。心臓、筋肉、脳、肺、腸などの一部の組織の特殊性により、分子分析用の高品質の核の産生を保証するために、各タイプのサンプルに適合した組織破壊および核分離の方法が必要です7,8,9,10 .組織破壊法および解離プロトコルは、機械的、酵素的(例えば、コラゲナーゼとDNaseの混合物)、または2つの組み合わせであり、手動または器具(例えば、Qiagen DSC-400、gentleMACS)によって行うことができる。
シングルセル技術は、生物医学研究に最適なツールとなっています。神経生物学では、脳内の細胞の多様性とその機能の複雑さにより、希少な細胞集団を可視化し、それらの不均一性を評価するために、高解像度およびハイスループット分析が必要です11,12,13,14。細胞の同一性と個々の細胞の遺伝子制御機構を結びつけることで、脳の発達と生理学に関する洞察が得られます。別の例としては、感染性疾患、自己免疫疾患、または癌性疾患における免疫応答の研究があり、これは単一細胞解析に強く依存しています。免疫細胞サブセットの不均一性、その活性の複雑さ、他の細胞タイプとの相互作用により、免疫応答の根底にあるメカニズムを解読するには、単一細胞の分解能が必要です。免疫細胞は骨髄に由来し、造血前駆細胞は徐々に分化する細胞で構成されており、骨髄から末梢の家に出る前に、段階的なプロセスを通じて細胞表面マーカーを獲得および喪失します。シングルセル解析により、細胞の発生段階を詳細に評価することができます。これは、従来マルチパラメーターフローサイトメトリーによって行われていたシングルセル表現型によって達成できます。しかしながら、単一細胞トランスクリプトームシグネチャーは、これらの細胞が互いに分類されるクラスターに分布しており、したがって粗い細胞表面マーカーアプローチを使用するときに誤認される可能性があるため、前駆細胞サブタイプのより正確な同定を明らかにすることが示されている15。造血幹細胞および前駆細胞(HSPC)がさまざまな薬剤への曝露から獲得できるエピジェネティックな修飾を明らかにする研究が増えており、免疫系の長期的な応答性に大きな影響を与えています16,17,18,19。新しいマルチオミクス技術により、これらのプロセスをシングルセル分解能で研究することができます。
細胞および核の分離のための多くのプロトコルは頭脳11,20,21,22および骨髄のサンプル23,24のために記述されている。実験のばらつきによるバイアスを最小限に抑えるには、単一細胞トランスクリプトームおよびエピゲノムシーケンシングの結合用に最適化された単一核調製プロトコルを検証し、それによって単一細胞マルチオミクスアッセイの再現性を確保する必要があります。
ここでは、(1)新鮮凍結脳組織および(2)下流のシングルセルマルチオーム解析用の新鮮骨髄HSPCからの核調製のための2つの堅牢なプロトコルについて説明します(図1)。
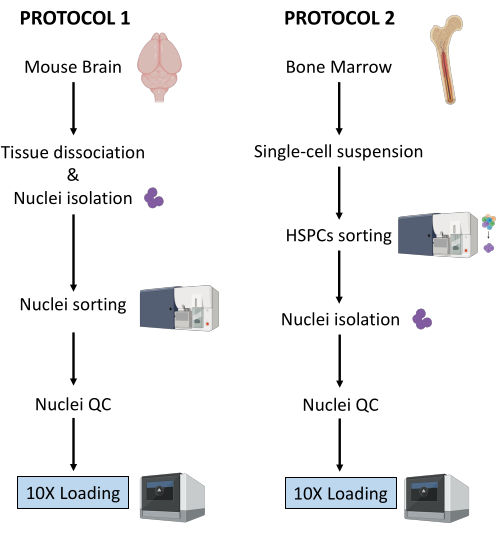
図1:新鮮凍結脳および骨髄組織からの核単離のプロトコルの概略図。 この図の拡大版をご覧になるには、ここをクリックしてください。
高品質の細胞または核懸濁液の調製は、単一細胞または単一核のRNA-Seqおよび単一細胞のマルチオミクス解析の成功にとって非常に重要です29,30,31。ここでは、脳と骨髄の2種類の組織からのマルチオームアッセイのためのサンプル調製と核単離のプロトコルについて説明しました。
この論文で説明されている脳プロトコルは、新鮮凍結された脳組織から高品質の核を回収することを可能にします。これには、凍結組織破壊、核の単離、核の精製、および調製された材料の品質管理のステップが含まれます。脳組織は多くの異なる種類の細胞で構成されており、組織の解離と核の分離の手順により、初期組織に存在する細胞集団の割合が維持されるはずです。ここでは、溶解バッファーの組成とインキュベーション時間を最適化して、組織を構成するすべての細胞集団を完全かつ穏やかに溶解できるようにしました。
骨髄HSPCのプロトコルは、実験の開始時に、不均一な細胞懸濁液から目的の細胞集団を単離するために1つの追加ステップを必要とするため、多少異なります。新鮮な組織を採取した後、赤血球を溶解し、サンプルを目的の細胞サブセットに濃縮します。標的細胞を溶解し、核を単離し、調製した材料の品質を制御します。
10X Genomicsは、多数の異なる組織における核分離について検証されたいくつかのプロトコルを提供する32,33。同社はまた、検証された組織から核を単離するための簡単なパイプラインを備えた核分離キットを商品化している34。ただし、これらのプロトコルでは、特定のサンプルの特殊性を調整するために、追加の最適化が必要です。たとえば、低いセル入力で作業する必要があるサンプルです。これらのサンプルの場合、最も困難なステップは遠心分離であり、サンプルを洗浄するのに十分な厳格さと、細胞/核の損失を回避するのに十分なほど穏やかな遠心分離が必要です。ここで説明するプロトコルでは、10X Genomics Demonstrated Protocol – Nuclei Isolation for Single Cell Multiome ATAC + GEX Sequencing (CG000365 – Rev C)27 を適応させ、これら 2 つの要件の微妙なバランスを見つけました。選別したHSPCから核を作製した例で示したように、サンプルの品質に影響を与えることなく、核の回収率を向上させました。
さらなる課題は、核単離のための精製細胞の溶解のステップです。より厳しい溶解条件と長いインキュベーション時間は、核を損傷し、それによってシーケンシングデータの品質に影響を与える可能性があります。 図5 は、溶解バッファーとの異なるインキュベーション時間における骨髄サンプルからの代表的な核イメージングを示しており、細胞溶解によって核の状態がどのように異なるかを示しています。HSPCの例では、健康そうに見える無傷の核の割合が最も高く、損傷した核の割合が最も低くなる状態として、3分間の溶解を特定しました。溶解インキュベーション時間は、新しいタイプのサンプルごとに最適化する必要があります。
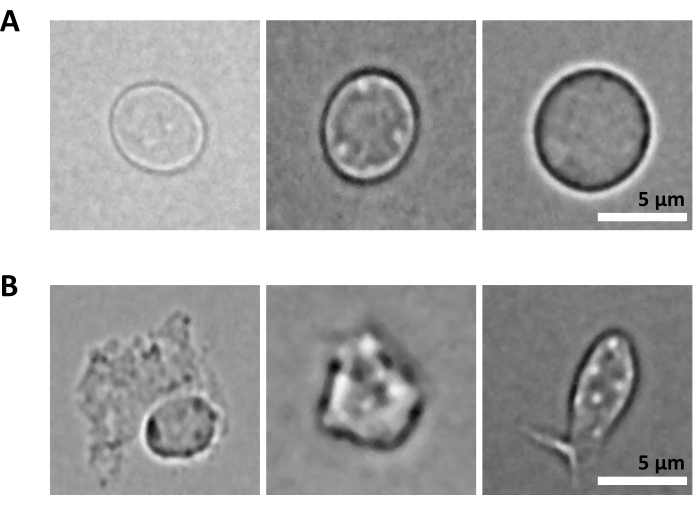
図5:顕微鏡による核の品質管理。 (A)無傷の核と(B)損傷した核を持つマウス骨髄から単離された核の代表的な明視野画像を示します。スケールバー5μm。画像は倒立顕微鏡で撮影し、40倍ELWD NA 0.60対物レンズと1.5倍デジタルズームを使用しました。 この図の拡大版をご覧になるには、ここをクリックしてください。
この作業で詳述されている両方のプロトコルは、ハイスループットFACS装置による標的細胞または核の精製に依存しています。このステップは、細胞のまれなサブセットを不均一な懸濁液から単離する単一細胞/核調製プロトコルにとって非常に重要です。これらのケースでは、HSPCのソーティングに示した例のように、目的の細胞集団の「ゲーティング」を可能にするために、高次元のフローサイトメトリーパネルが必要になる場合があります。選別は非常に高速かつ正確で、選別された細胞サブセットの純度は95%を超えます。このアプローチは、細胞懸濁液を最大70psiの圧力にさらすため、細胞膜の破裂を引き起こす可能性があるため、脆弱な細胞(樹状細胞、好中球など)の選別を制限する可能性があります。このような場合、細胞精製には、磁気選別、新世代機器(CellenOne、Cellenionなど)の適用など、代替溶液を選択する必要があります。MACSQuant Tyto, Miltenyi)35,36、または液滴ベースのシステム(ODIN、Sensificなど)37。それにもかかわらず、これらの技術のソーティング速度は遅く、細胞の選別は数分ではなく数時間続くため、多数の細胞の解析に基づくMultiomeやその他のシングルセルアプリケーション用の生細胞の調製にこれらのアプローチを適用する際の強力な制限要因となっています。
組織から単離された核の精製には、そのスループットと単離された材料の純度から、FACSが最適な方法です。核は圧力に敏感ではなく、ろ過された組織分離株はセルソーターで簡単に精製できます。実験室にFACS機器が装備されていない場合は、他の代替手段が存在し、やや効率は劣りますが、十分に優れています。例としては、超遠心分離や、音波を使用して粒子のサイズの違いに基づいて粒子を分離するMARS(アプライドセル)などの小型機器の使用が含まれます。細胞/核懸濁液の疎水性を利用したCURIOX層流洗浄機。または、細胞の物理的特性(浮遊)に依存して細胞を破片から分離するLEVITAS bio。
ここでは、ダウンストリームのMultiomeプロトコルに多数の核と最適な純度を得るためのプロトコルについて説明します。FACSによる選別と遠心分離の繰り返しは、最初の材料の大幅な損失をもたらします。このため、ここで説明する脳からの核調製プロトコルでは、FACSソーティング後に少なくとも500,000個の核を収集するのに十分な量の出発物質が必要です。この基準が一致しない場合は、代替プロトコルを適用する必要があります。稀な細胞集団や小さな組織切片を扱う場合、利用可能な初期物質の量が制限要因となる可能性があります。この問題に対処するために、(a)溶解量を減らす、(b)洗浄量を減らす、(c)遠心分離時間を延長した1回の洗浄で回収率を向上させることで、低細胞入力核単離のための10X Genomicsプロトコルに示されているように、核の回収率を向上させることができます。低含有量材料のマルチオミクス解析には、scNMT、SNARE-seq、Paired-seq38 など、必要なインプットサンプルがはるかに少ないプレートベースのアプリケーションを検討する価値があります。
要約すると、ダウンストリームマルチオーム解析用の脳および骨髄HSPCからの核調製のための2つの堅牢なプロトコルについて説明しました。これらのプロトコルは、提起された科学的な問題に関係なく、これら2種類の組織から高品質の単核懸濁液を必要とするあらゆる科学プロジェクトに適用できます。私たちのグループでは、脳核単離プロトコルを、様々な標的遺伝子の不活性化による脳の発生研究や、神経疾患における免疫応答の研究に応用しています。私たちは、骨髄核単離プロトコルを使用して、免疫系の確立におけるさまざまな造血亜集団の関与を解読しています。
