Bioprinting tridimensionale di cocolture neurone-astrociti derivate da iPSC umane per applicazioni di screening
Summary
Qui, presentiamo un protocollo per produrre cocolture biostampate in 3D di neuroni e astrociti derivati da iPSC. Questo modello di cocoltura, generato all’interno di un’impalcatura di idrogel in formati a 96 o 384 pozzetti, dimostra un’elevata vitalità post-stampa e la crescita dei neuriti entro 7 giorni e mostra l’espressione di marcatori di maturità per entrambi i tipi di cellule.
Abstract
Affinché un modello cellulare sia utilizzabile per lo screening farmacologico, il sistema deve soddisfare i requisiti di produttività e omogeneità, oltre ad avere un tempo di sviluppo efficiente. Tuttavia, molti modelli 3D pubblicati non soddisfano questi criteri. Questo, quindi, limita la loro utilità nelle prime applicazioni di scoperta di farmaci. La biostampa tridimensionale (3D) è una nuova tecnologia che può essere applicata allo sviluppo di modelli 3D per accelerare i tempi di sviluppo, aumentare la standardizzazione e aumentare la produttività. Qui, presentiamo un protocollo per sviluppare modelli di cocoltura biostampati in 3D di neuroni e astrociti glutammatergici derivati da cellule staminali pluripotenti indotte umane (iPSC). Queste cocolture sono incorporate all’interno di una matrice idrogel di peptidi bioattivi, proteine della matrice extracellulare (ECM) a lunghezza intera e con una rigidità fisiologica di 1,1 kPa. Il modello può essere rapidamente stabilito nei formati a 96 e 384 pozzetti e produce una vitalità media post-stampa del 72%. Il rapporto astrocita/neurone in questo modello è di 1:1,5, che rientra nell’intervallo fisiologico per il cervello umano. Queste popolazioni di cellule biostampate in 3D mostrano anche l’espressione di marcatori di tipo di cellule neurali mature e la crescita delle proiezioni di neuriti e astrociti entro 7 giorni dalla coltura. Di conseguenza, questo modello è adatto per l’analisi utilizzando coloranti cellulari e tecniche di immunocolorazione insieme a saggi di crescita neuritica. La capacità di produrre questi modelli fisiologicamente rappresentativi su larga scala li rende ideali per l’uso in saggi di screening a medio-alto rendimento per bersagli neuroscientifici.
Introduction
La ricerca sulle malattie del sistema nervoso centrale (SNC) nel settore della scoperta di farmaci si sta espandendo1. Tuttavia, molte malattie prevalenti del sistema nervoso centrale come l’epilessia, la schizofrenia e il morbo di Alzheimer non hanno ancora trattamenti curativi 2,3,4. La mancanza di terapie efficaci nelle malattie del sistema nervoso centrale può, almeno in parte, essere attribuita alla mancanza di modelli in vitro accurati del cervello5. Ciò ha comportato un divario traslazionale tra gli attuali modelli in vitro e i dati in vivo e un conseguente collo di bottiglia negli sforzi di ricerca.
Spinto da questo divario traslazionale, negli ultimi anni c’è stato un aumento significativo nello sviluppo di nuovi modelli cellulari 3D, tra cui organoidi neurali, neurosferoidi e modelli basati su scaffold6. La struttura 3D di questi modelli aiuta a ricapitolare il microambiente neurale, compresi gli stress biomeccanici, i contatti cellula-cellula e la matrice extracellulare cerebrale (ECM)7. L’ECM cerebrale è un elemento dinamico della neurofisiologia che occupa lo spazio tra i tipi di cellule neurali, tra cui neuroni, astrociti, oligodendrociti e l’unità neurovascolare7. È stato dimostrato che la ricapitolazione dell’ECM cerebrale influenza la morfologia neuronale e l’attivazione neuronale e molti modelli 3D complessi del cervello hanno dimostrato la deposizione di proteine ECM da parte dei tipi di cellule neurali 8,9,10,11. I modelli basati su scaffold sono costituiti da cocolture neurali mature sospese in una matrice porosa di idrogel sintetico o biologico che rappresenta l’ECM12 del cervello. A differenza dei sistemi organoidi e sferoidi, i modelli 3D basati su scaffold consentono la personalizzazione delle proteine ECM presenti e hanno l’ulteriore vantaggio della sintonizzabilità della rigidità dell’idrogel per imitare le sollecitazioni biomeccaniche13,14.
Sebbene la stragrande maggioranza dei modelli neurali 3D dimostri una maggiore ricapitolazione del microambiente cerebrale, non tutti i modelli sono adatti per l’implementazione di applicazioni di scoperta farmacologica15. Affinché un modello 3D possa essere implementato nei processi industriali, il sistema deve soddisfare i requisiti di produttività per le applicazioni di screening e avere un tempo di sviluppo relativamente breve16. La biostampa 3D è una nuova tecnologia che offre il potenziale per creare modelli neurali basati su scaffold 3D con tempi di sviluppo rapidi, maggiore produttività e livelli più elevati di controllo di precisione, oltre alla rimozione della variabilità causata dall’errore umano17. Questo protocollo presenta un modello di cocoltura 3D di neuroni glutammatergici e astrociti umani derivati da iPSC in un’impalcatura di idrogel. Questa impalcatura in idrogel contiene peptidi bioattivi fisiologicamente rappresentativi (RGD, IKVAV, YIGSR) e proteine ECM all’interno di una rigidità biomeccanica mimetica. Queste proteine ECM a lunghezza intera includono laminina-211 a lunghezza intera e acido ialuronico, abbondanti nella corteccia umana, con una rigidità di 1,1 kPa in linea con le misurazioni in vivo 18. Questo modello è progettato con praticità per la scoperta di farmaci ed è creato utilizzando una biostampante 3D in un formato di piastra a 96 pozzetti o 384 pozzetti adatto per l’analisi di screening utilizzando tecniche di imaging con coloranti cellulari e anticorpi, insieme a saggi di crescita neuritica. Le cellule mostrano l’espressione di marcatori di tipo di cellula neurale e la crescita di proiezioni neuritiche e astrocitarie entro 7 giorni dalla coltura. Pertanto, questo protocollo presenterà la metodologia per sviluppare un modello di cocoltura neurale 3D ad alto rendimento da utilizzare in applicazioni di scoperta di farmaci.
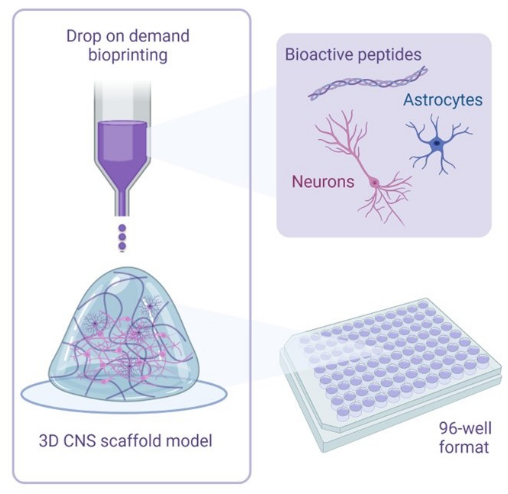
Figura 1: Panoramica illustrativa della metodologia utilizzata per le cocolture di biostampa 3D. I neuroni e gli astrociti umani derivati da iPSC sono combinati con soluzioni attivatrici e bioink contenenti peptidi bioattivi e sono biostampati su scaffold di idrogel in formati a 96 o 384 pozzetti utilizzando la tecnologia di bioprinting drop-on-demand. Fare clic qui per visualizzare una versione più grande di questa figura.
Protocol
Representative Results
Discussion
La necessità di modelli accurati del sistema nervoso centrale non è mai stata così elevata e i limiti dei modelli di coltura cellulare tradizionali bidimensionali (2D) hanno portato a una generazione di modelli complessi del sistema nervoso centrale negli ultimi anni19. Tuttavia, molti modelli 3D complessi che rappresentano le interazioni tra i tipi di cellule neurali e l’ECM hanno limitazioni che impedirebbero l’applicazione di questi modelli nei processi industriali 6,20,21. In questo protocollo, sviluppiamo un modello di cocoltura 3D di neuroni e astrociti umani derivati da iPSC, che mira a risolvere alcune di queste limitazioni utilizzando la tecnologia di bioprinting 3D per creare un’impalcatura di idrogel bioattivo nei formati a 96 e 384 pozzetti.
La metodologia per lo sviluppo di questi modelli è stata semplificata attraverso il software di progettazione della mappa delle lastre, i protocolli di stampa generati automaticamente e il processo di stampa guidato dalla biostampante. Tuttavia, a causa della natura sensibile dei tipi di cellule sensibili derivate da iPSC utilizzati in questo protocollo, è necessario prestare attenzione alle seguenti fasi critiche dello scongelamento e della coltura. In primo luogo, l’inclusione dell’inibitore ROCK (ROCKi) ha diversi vantaggi durante il processo di bioprinting e durante la coltura precoce. Lo scongelamento cellulare è un punto critico in cui i neuroni possono sperimentare una risposta allo stress e protocolli di scongelamento impropri possono ridurre le possibilità di sopravvivenza22. In genere si consiglia di scongelare le cellule, aggiungere terreni e portare le cellule alla temperatura dell’incubatrice nel modo più efficiente possibile23. Tuttavia, durante il processo di bioprinting descritto in questo protocollo, è necessario che i neuroni e gli astrociti siano risospesi in una soluzione attivatrice piuttosto che in un supporto, e le cellule non saranno sollevate al di sopra della temperatura ambiente fino alla fine della tiratura (fino a 30 minuti dopo il disgelo). Pertanto, l’aggiunta di ROCKi al terreno immediatamente dopo lo scongelamento e l’inclusione di questo durante le due fasi di centrifugazione (fasi 2.1–2.7 e 1.3.15-1.3.20) è indispensabile per inibire le vie di stress cellulare, che si tradurrebbero in livelli di vitalità più bassi24. Inoltre, è stato dimostrato che ROCKi promuove la crescita dei neuriti e migliora la maturazione neuronale25. Pertanto, l’integrazione di ROCKi viene continuata per 48 ore dopo il bioprinting. Tuttavia, è imperativo rimuovere l’integrazione di ROCKi dopo 48 ore per garantire il lavaggio completo durante i successivi cambi di terreno prima che le cellule vengano utilizzate per il saggio.
Un’ulteriore fase che richiede un’attenzione critica è durante l’aggiunta e la sostituzione dei supporti post-stampa (passaggi 2.8-2.13). Lo scaffold in idrogel biostampato ha una rigidità biomeccanica equivalente di soli 1,1 kPa, equivalente alla materia grigia. Come descritto al punto 2.10, è fondamentale pipettare delicatamente nel lato del pozzetto durante l’aggiunta e l’aspirazione del terreno per evitare disturbi. Ciò è particolarmente rilevante per le piastre a 384 pozzetti, dove il livello di gel occupa una percentuale maggiore del volume totale del pozzetto. Questo metodo dovrebbe essere utilizzato anche nei pozzetti di controllo 2D per prevenire il sollevamento dei bordi delle cellule e il taglio delle escrescenze di neuriti. Gli autori vorrebbero anche sottolineare l’importanza della tecnica sterile all’interno della bioprinter, che dovrebbe essere trattata con una cautela equivalente a quella di una cabina di biosicurezza utilizzata per le colture cellulari derivate da iPSC. Ciò include il filtraggio sterile del 70% di EtOH e dH2O utilizzato nelle procedure di illuminazione e stampa, il mantenimento dei coperchi sulle cartucce e sulle piastre mentre si muovono le mani dentro e fuori dalla biostampante e la decontaminazione delle superfici all’interno della biostampante con salviette al 70% di etanolo prima e dopo la stampa.
L’impalcatura di idrogel biostampata, formata da soluzioni di bioink e attivatori, selezionata per sviluppare questo modello è selezionata da una gamma di bioink e soluzioni attivatrici sviluppate da Inventia Life Science per l’uso all’interno della biostampante RASTRUM. La laminina e l’acido ialuronico sono stati identificati come molecole rilevanti per la maturazione neuronale derivata da iPSC a causa del loro ruolo nella guida assonale, nella formazione delle sinapsi e nella formazione della rete perineuronale26,27. Inoltre, è stata selezionata una rigidità biomeccanica di 1,1 kPa, poiché è stato dimostrato che gli idrogel a bassa densità consentono una migliore crescita dei neuriti dai neuroni12. Se vengono apportate modifiche al protocollo utilizzando neuroni e astrociti che sono stati differenziati internamente o da un diverso fornitore commerciale, si consiglia di eseguire un test di selezione della matrice per determinare l’impalcatura di idrogel più favorevole15. Inoltre, potrebbe anche essere necessario ottimizzare la densità cellulare se vengono apportate modifiche alle fonti cellulari per garantire una vitalità ottimale e prevenire il sovraffollamento dell’idrogel. Per tutte le modifiche e la risoluzione dei problemi relativi alla funzione bioprinter, gli autori raccomandano di contattare i produttori e di fare riferimento ai protocolli del produttore.
Il sistema nervoso centrale contiene un’ampia gamma di sottotipi neuronali e cellule gliali, che esistono tutti in diverse nicchie cerebrali e hanno ruoli specifici che contribuiscono alla funzione neurale28. Nel contesto di questo ampio ambito, questo modello rappresenta solo i due tipi cellulari più abbondanti (astrociti e neuroni glutammatergici eccitatori). Importanti tipi di cellule come la microglia, gli oligodendrociti e le cellule endoteliali che formano la barriera emato-encefalica sono omessi da questo sistema. L’inclusione della microglia potrebbe essere rilevante per concentrarsi sulle interazioni neuroimmunitarie e gli oligodendrociti potrebbero essere di interesse nelle malattie che colpiscono la mielinizzazione centrale. Oltre al loro ruolo nella patologia, cellule come le cellule endoteliali che formano la barriera emato-encefalica espellono enzimi che metabolizzano i farmaci, il che potrebbe influenzare l’uso di questo modello per i saggi di farmacocinetica29. Un’ulteriore limitazione del modello potrebbe essere il rapporto tra astrociti e neuroni; Il rapporto tra astrociti e neuroni varia notevolmente tra le regioni del cervello, con valori suggeriti compresi tra 1:1 e 1:330,31. Questo modello ha un rapporto approssimativo di 1:1,5 astrociti rispetto ai neuroni; Pertanto, questo modello potrebbe non essere rilevante per le aree cerebrali modello in cui gli astrociti sono più abbondanti, come nelle aree della sostanza bianca30.
Negli ultimi anni sono stati pubblicati altri protocolli per sviluppare modelli di cocoltura biostampati in 3D. Una pubblicazione di Sullivan et al., 2021, ha presentato un modello neurale biostampato in 3D utilizzando cellule progenitrici neurali derivate da iPSC, che dimostra un’elevata vitalità post-stampa e un miglioramento della funzione neuronale rispetto alle colture 2D32. Tuttavia, in questo protocollo, le cellule progenitrici neurali sono state utilizzate come fonte di cellule e sono state mantenute in coltura per 4 settimane. In questo protocollo, sono stati utilizzati neuroni glutammatergici e astrociti derivati da iPSC disponibili in commercio. Ciò consente di creare una rete 3D di cellule co-coltivate in soli 7 giorni; Come dimostrato dall’analisi della crescita dei neuriti, la crescita dei neuriti inizia entro 24 ore e continua in modo lineare per tutto il periodo di 156 ore per il quale è stata monitorata la crescita cellulare. La rapida costituzione di queste reti può essere in parte attribuita all’uso di neuroni glutammatergici che utilizzano l’espressione genica indotta dalla doxiciclina ottimizzata di NGN2, che mostra l’espressione di marcatori di sottotipo neuronale maturi entro 7 giorni, anche in coltura 2D33. L’accorciamento di questo periodo di crescita con questa tecnica è importante per l’implementazione di modelli all’interno dell’industria biofarmaceutica, poiché lo sviluppo di saggi richiede una rapida inversione di tendenza e lo sviluppo di modelli cellulari15.
In conclusione, questo modello mostra il potenziale per un modello 3D di neuroni e astrociti, che viene stabilito in modo rapido e conveniente per scopi di screening. Le applicazioni future per questo tipo di modello potrebbero riguardare gli sforzi di scoperta di farmaci in diverse malattie del SNC, con l’opportunità di espandersi a diverse malattie utilizzando linee iPSC di pazienti o malattie geneticamente modificate. Inoltre, l’uso di neuroni glutammatergici derivati da iPSC inducibili dalla doxiciclina NGN2 consente alle cellule di raggiungere la maturità in meno tempo, che può essere utilizzato per lo sviluppo di modelli del cervello che invecchia per la ricerca sulla neurodegenerazione. Questo sistema potrebbe anche essere ampliato attraverso l’uso di ulteriori tipi di cellule in cocoltura, tra cui microglia e oligodendrociti.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Gli autori desiderano ringraziare Alex Volkerling, Martin Engel e Rachel Bleach per la loro assistenza nello sviluppo del protocollo e per il feedback sul manoscritto.
Materials
| 2-mercaptoethanol | Thermofisher | 31350010 | |
| 384-well plate | PerkinElmer | 6057300 | |
| 96-well plate | PerkinElmer | 6055300 | |
| Activator fluid F299 | Inventia Life Science | N/A | |
| Activator fluid F3 | Inventia Life Science | N/A | |
| B27 (50x) minus Vit A | Thermofisher | 12587010 | |
| Bioink fluid F261 | Inventia Life Science | N/A | |
| Bioink fluid F32 | Inventia Life Science | N/A | |
| Doxycycline hyclate | Sigma Aldrich | D5207 | |
| GlutaMAX (100x) | Thermofisher | 35050061 | |
| Goat anti-mouse IgG H&L Alexa Fluor 647 | Abcam | ab150115 | |
| Goat anti-rabbit IgG H&L Alexa Fluor 488 | Abcam | ab150077 | |
| Hoechst | Abcam | ab228551 | |
| Human BDNF Recombinant Protein | Thermofisher | PHC7074 | |
| Human NT3 Recombinant Protein | Thermofisher | PHC7036 | |
| iCell Astrocytes | Fujifilm CDI | 1434 | |
| INCell Analyser 6500HS | Molecular Devices | N/A | high content imaging system |
| Incucyte S3 | Sartorius | N/A | |
| ioGlutamatergic Neurons (Large vial) | Bit.bio | e001 | |
| Laminin (1 mg/mL) | Sigma Aldrich | L2020 | |
| Live/dead kit (Calcein-AM, Ethidium homo-dimer-1) | Invitrogen | L3224 | |
| Mouse anti-BIII tubulin NL637 conjugated | R&D systems | SC024 | |
| Neurobasal media | Thermofisher | 21103049 | |
| Normal Donkey Serum | Abcam | ab7475 | |
| NucBlue Live (Hoechst 33342) | Thermofisher | R37605 | |
| Opti-MEM | Thermofisher | 11058021 | |
| Paraformaldehyde | Sigma Aldrich | P6148 | |
| PEI 50% in H2O | Sigma Aldrich | 181978 | |
| Pierce Borate Buffer 20x | Thermofisher | 28341 | |
| Prism | GraphPad | Data analysis software | |
| Rabbit anti-ionotropic glutamatre receptor 2 (GluR2) | Abcam | ab206293 | |
| RASTRUM(TM) Bioprinter | Inventia Life Science | N/A | Bioprinter |
| RASTRUM(TM) Bioprinter Cartridges | Inventia Life Science | N/A | Bioprinter Cartridges |
| RASTRUM(TM) Targeting plate | Inventia Life Science | N/A | Targeting plate |
| Rho kinase (ROCK) inhibitor | Abcam | ab120129 | |
| Sheep anti-GFAP NL493 conjugated | R&D systems | SC024 | |
| Signals Image Artist | PerkinElmer | N/A | Image analysis platform |
| Triton X-100 | Thermofisher | HFH10 | |
| Zeiss Axio Observer | Zeiss | N/A | Inverted microscope platform |
References
- Jung, Y. L., Hwang, J., Yoo, H. S. Disease burden metrics the innovations of leading pharmaceutical companies: a global and regional comparative study. Globalization and Health. 16 (1), 80-80 (2020).
- Potkin, S. G., et al. The neurobiology of treatment-resistant schizophrenia: paths to antipsychotic resistance and a roadmap for future research. npj Schizophrenia. 6, 1 (2020).
- Zahra, W., et al., Keswani, C., et al. The Global Economic Impact of Neurodegenerative Diseases: Opportunities and Challenges. Bioeconomy for Sustainable Development. , (2019).
- Perucca, E. The pharmacological treatment of epilepsy: recent advances and future perspectives. Acta Epileptologica. 3 (1), 22 (2021).
- Nikolakopoulou, P., et al. Recent progress in translational engineered in vitro models of the central nervous system. Brain. 143 (11), 3181-3213 (2020).
- Whitehouse, C., Corbett, N., Brownlees, J. 3D models of neurodegeneration: implementation in drug discovery. Trends in Pharmacological Sciences. 44 (4), 208-221 (2023).
- Rauti, R., Renous, N., Maoz, B. M. Mimicking the brain extracellular matrix in vitro: A review of current methodologies and challenges. Israel Journal of Chemistry. 60 (12), 1141-1151 (2020).
- Fawcett, J. W., Oohashi, T., Pizzorusso, T. The roles of perineuronal nets and the perinodal extracellular matrix in neuronal function. Nature Reviews Neuroscience. 20 (8), 451-465 (2019).
- Lam, D., et al. Tissue-specific extracellular matrix accelerates the formation of neural networks and communities in a neuron-glia co-culture on a multi-electrode array. Scientific Reports. 9, 4159 (2019).
- Roll, L., Lessmann, K., Brüstle, O., Faissner, A. Cerebral organoids maintain the expression of neural stem cell-associated glycoepitopes and extracellular matrix. Cells. 11 (5), 760 (2022).
- Yan, Y., Bejoy, J., Marzano, M., Li, Y. The use of pluripotent stem cell-derived organoids to study extracellular matrix development during neural degeneration. Cells. 8 (3), 242 (2019).
- Ma, L., et al. 3D bioprinted hyaluronic acid-based cell-laden scaffold for brain microenvironment simulation. Bio-Design and Manufacturing. 3 (3), 164-174 (2020).
- Liaw, C. -. Y., Ji, S., Guvendiren, M. Engineering 3D hydrogels for personalized in vitro human tissue models. Advanced Healthcare Materials. 7 (4), 1701165 (2018).
- Ma, J., Huang, C. Composition and mechanism of three-dimensional hydrogel system in regulating stem cell fate. Tissue Engineering Part B: Reviews. 26 (6), 498-518 (2020).
- Belfiore, L., et al. Generation and analysis of 3D cell culture models for drug discovery. European Journal of Pharmaceutical Sciences. 163, 105876 (2021).
- Langhans, S. A. Three-dimensional in vitro cell culture models in drug discovery and drug repositioning. Frontiers in Pharmacology. 9, 6 (2018).
- Engel, M., Belfiore, L., Aghaei, B., Sutija, M. Enabling high throughput drug discovery in 3D cell cultures through a novel bioprinting workflow. SLAS Technology. 27 (1), 32-38 (2022).
- Takamura, T., et al. Influence of age on global and regional brain stiffness in young and middle-aged adults. Journal of Magnetic Resonance Imaging. 51 (3), 727-733 (2020).
- Slanzi, A., Iannoto, G., Rossi, B., Zenaro, E., Constantin, G. In vitro models of neurodegenerative diseases. Frontiers in Cell and Developmental Biology. 8, 328 (2020).
- de Souza, N. Organoid variability examined. Nature Methods. 14 (7), 655-655 (2017).
- Hernández, D., et al. Culture variabilities of human iPSC-derived cerebral organoids are a major issue for the modelling of phenotypes observed in Alzheimer’s disease. Stem Cell Review and Reports. 18 (2), 718-731 (2022).
- Li, R., et al. Differentiation of human iPS cells into sensory neurons exhibits developmental stage-specific cryopreservation challenges. Frontiers in Cell and Developmental Biology. 9, 796960 (2021).
- Nishiyama, Y., et al. Safe and efficient method for cryopreservation of human induced pluripotent stem cell-derived neural stem and progenitor cells by a programmed freezer with a magnetic field. Neuroscience Research. 107, 20-29 (2016).
- Uhrig, M., Ezquer, F., Ezquer, M. Improving cell recovery: Freezing and thawing optimization of induced pluripotent stem cells. Cells. 11 (5), 799 (2022).
- Harbom, L. J., et al. The effect of rho kinase inhibition on morphological and electrophysiological maturity in iPSC-derived neurons. Cell and Tissue Research. 375 (3), 641-654 (2019).
- Koh, H. S., Yong, T., Chan, C. K., Ramakrishna, S. Enhancement of neurite outgrowth using nano-structured scaffolds coupled with laminin. Biomaterials. 29 (26), 3574-3582 (2008).
- Tarus, D., et al. Design of hyaluronic acid hydrogels to promote neurite outgrowth in three dimensions. ACS Applied Materials & Interfaces. 8 (38), 25051-25059 (2016).
- Brain Initiative Cell Census Network (BICCN). Initiative Cell Census Network (BICCN). A multimodal cell census and atlas of the mammalian primary motor cortex. Nature. 598 (7879), 86-102 (2021).
- Dauchy, S., et al. Expression and transcriptional regulation of ABC transporters and cytochromes P450 in hCMEC/D3 human cerebral microvascular endothelial cells. Biochemical Pharmacology. 77 (5), 897-909 (2009).
- Herculano-Houzel, S. The glia/neuron ratio: How it varies uniformly across brain structures and species and what that means for brain physiology and evolution. Glia. 62 (9), 1377-1391 (2014).
- von Bartheld, C. S., Bahney, J., Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. The Journal of Comparative Neurology. 524 (18), 3865-3895 (2016).
- Sullivan, M. A., et al. 3D bioprinting of stem cell-derived central nervous system cells enables astrocyte growth, vasculogenesis and enhances neural differentiation/function. bioRxiv. , (2022).
- Pawlowski, M., et al. Inducible and deterministic forward programming of human pluripotent stem cells into neurons, skeletal myocytes, and oligodendrocytes. Stem Cell Reports. 8 (4), 803-812 (2017).
