Bioimpressão Tridimensional de Coculturas Neurônio-Astrócitos Derivadas de iPSC Humanas para Aplicações de Triagem
Summary
Aqui, apresentamos um protocolo para produzir coculturas bioimpressas em 3D de neurônios e astrócitos derivados de iPSC. Este modelo de cocultura, gerado dentro de um arcabouço de hidrogel em formatos de 96 ou 384 poços, demonstra alta viabilidade pós-impressão e crescimento de neuritos em 7 dias e mostra a expressão de marcadores de maturidade para ambos os tipos celulares.
Abstract
Para que um modelo de célula seja viável para a triagem de drogas, o sistema deve atender aos requisitos de produtividade e homogeneidade, além de ter um tempo de desenvolvimento eficiente. No entanto, muitos modelos 3D publicados não satisfazem esses critérios. Isso, portanto, limita sua utilidade em aplicações de descoberta precoce de drogas. A bioimpressão tridimensional (3D) é uma nova tecnologia que pode ser aplicada ao desenvolvimento de modelos 3D para agilizar o tempo de desenvolvimento, aumentar a padronização e aumentar o rendimento. Aqui, apresentamos um protocolo para desenvolver modelos de cocultura bioimpressos 3D de neurônios glutamatérgicos e astrócitos derivados de células-tronco pluripotentes induzidas por humanos (iPSC). Essas coculturas estão embutidas dentro de uma matriz hidrogel de peptídeos bioativos, proteínas da matriz extracelular (MEC) de comprimento total e com rigidez fisiológica de 1,1 kPa. O modelo pode ser rapidamente estabelecido em formatos de 96 e 384 poços e produz uma viabilidade pós-impressão média de 72%. A relação astrócito-neurônio neste modelo é mostrada como sendo 1:1,5, que está dentro da faixa fisiológica para o cérebro humano. Essas populações de células bioimpressas em 3D também mostram expressão de marcadores do tipo de células neurais maduras e crescimento de projeções de neuritos e astrócitos dentro de 7 dias de cultivo. Como resultado, este modelo é adequado para análise usando corantes celulares e técnicas de imunomarcação ao lado de ensaios de crescimento de neuritos. A capacidade de produzir esses modelos fisiologicamente representativos em escala os torna ideais para uso em ensaios de triagem de médio a alto rendimento para alvos de neurociência.
Introduction
A pesquisa em doenças do sistema nervoso central (SNC) na indústria de descoberta de medicamentos está em expansão1. No entanto, muitas doenças prevalentes do SNC, como epilepsia, esquizofrenia e doença de Alzheimer, ainda não apresentam tratamento curativo 2,3,4. A falta de terapêutica eficaz nas doenças do SNC pode, pelo menos em parte, ser atribuída à falta de modelos acurados in vitro do cérebro5. Isso resultou em uma lacuna translacional entre os modelos in vitro atuais e os dados in vivo e um subsequente gargalo nos esforços de pesquisa.
Impulsionado por essa lacuna translacional, houve um aumento significativo no desenvolvimento de novos modelos de células 3D nos últimos anos, incluindo organoides neurais, neuroesferoides e modelos baseados em arcabouços6. A estrutura 3D desses modelos auxilia na recapitulação do microambiente neural, incluindo estresses biomecânicos, contatos célula-célula e matriz extracelular cerebral (MEC)7. A MEC cerebral é um elemento dinâmico da neurofisiologia que ocupa o espaço entre os tipos de células neurais, incluindo neurônios, astrócitos, oligodendrócitos e a unidade neurovascular7. Demonstrou-se que a recapitulação da MEC cerebral afeta a morfologia neuronal e o disparo neuronal, e muitos modelos 3D complexos do cérebro demonstraram deposição de proteínas da MEC por tipos de células neurais 8,9,10,11. Os modelos baseados em scaffolds consistem em coculturas neurais maduras suspensas em uma matriz porosa sintética ou biológica de hidrogel que representa a MEC cerebral12. Ao contrário dos sistemas organoides e esferoides, os modelos 3D baseados em scaffolds permitem a personalização das proteínas da MEC presentes e têm o benefício adicional da tunabilidade da rigidez do hidrogel para mimetizar tensões biomecânicas13,14.
Embora a esmagadora maioria dos modelos neurais 3D demonstre uma maior recapitulação do microambiente cerebral, nem todos os modelos são adequados para implementar aplicações de descoberta de drogas15. Para que um modelo 3D seja implementado em processos industriais, o sistema deve atender aos requisitos de throughput para aplicações de triagem e ter um tempo de desenvolvimento relativamente curto16. A bioimpressão 3D é uma nova tecnologia que oferece o potencial de criar modelos neurais baseados em arcabouços 3D com rápido tempo de desenvolvimento, maior rendimento e níveis mais altos de controle de precisão, juntamente com a remoção da variabilidade causada pelo erro humano17. Este protocolo apresenta um modelo de cocultura 3D de neurônios glutamatérgicos e astrócitos humanos derivados da iPSC em um arcabouço de hidrogel. Este arcabouço de hidrogel contém peptídeos bioativos fisiologicamente representativos (RGD, IKVAV, YIGSR) e proteínas da MEC dentro de uma rigidez biomecânica mimética. Essas proteínas de MEC, de comprimento total, incluem laminina-211 e ácido hialurônico, abundantes no córtex humano, com rigidez de 1,1 kPa, de acordo com medidas in vivo 18. Este modelo é projetado com praticidade para a descoberta de drogas, e é criado usando uma bioimpressora 3D em um formato de placa de 96 ou 384 poços adequado para análise de triagem usando técnicas de imagem com corantes celulares e anticorpos, juntamente com ensaios de crescimento de neuritos. As células apresentam expressão de marcadores do tipo celular neural e crescimento de projeções neuríticas e astrocitárias em até 7 dias após o cultivo. Assim, este protocolo apresentará a metodologia para o desenvolvimento de um modelo de cocultura neural 3D de alto rendimento para uso em aplicações de descoberta de fármacos.
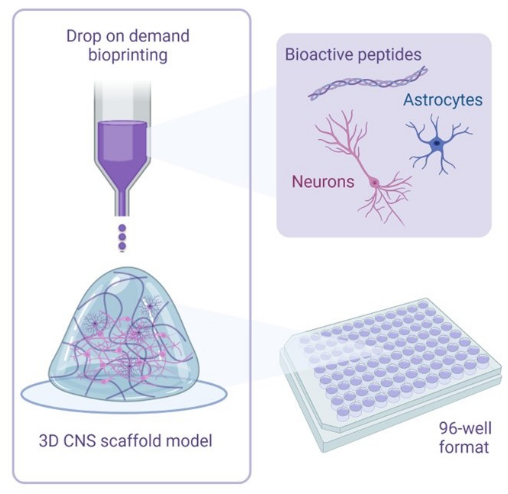
Figura 1: Visão geral ilustrativa da metodologia utilizada para coculturas de bioimpressão 3D. Neurônios e astrócitos humanos derivados de iPSC são combinados com soluções de ativador e biotinta contendo peptídeos bioativos e são bioimpressos em scaffolds de hidrogel em formatos de 96 ou 384 poços usando tecnologia de bioimpressão drop-on-demand. Clique aqui para ver uma versão maior desta figura.
Protocol
Representative Results
Discussion
A necessidade de modelos precisos do SNC nunca foi tão grande, e as limitações dos modelos tradicionais de cultura de células bidimensionais (2D) têm impulsionado uma geração de modelos complexos do SNC nos últimos anos19. Entretanto, muitos modelos 3D complexos que representam interações entre tipos de células neurais e a MEC apresentam limitações que impediriam a aplicação desses modelos em processos industriais 6,20,21. Neste protocolo, desenvolvemos um modelo de cocultura 3D de neurônios e astrócitos humanos derivados de iPSC, que visa resolver algumas dessas limitações usando a tecnologia de bioimpressão 3D para criar um arcabouço de hidrogel bioativo nos formatos de 96 e 384 poços.
A metodologia para o desenvolvimento desses modelos foi simplificada através do software de desenho de mapas de placas, protocolos de impressão gerados automaticamente e processo de impressão guiada a partir da bioimpressora. No entanto, devido à natureza sensível dos tipos celulares sensíveis derivados de iPSC usados neste protocolo, deve-se tomar cuidado com as seguintes etapas críticas no descongelamento e cultura. Em primeiro lugar, a inclusão do inibidor de ROCK (ROCKi) tem vários benefícios ao longo do processo de bioimpressão e durante o cultivo inicial. O descongelamento celular é um ponto crítico em que os neurônios podem experimentar uma resposta ao estresse, e protocolos de descongelamento inadequados podem diminuir as chances de sobrevivência22. Normalmente, recomenda-se descongelar células, adicionar meios e elevar as células à temperatura da incubadora da forma mais eficiente possível23. No entanto, durante o processo de bioimpressão descrito neste protocolo, é necessário que os neurônios e astrócitos sejam ressuspensos em uma solução ativadora em vez de meio, e as células não serão elevadas acima da temperatura ambiente até o final da tiragem (até 30 minutos pós-descongelamento). Assim, a adição de ROCKi ao meio imediatamente após o descongelamento e sua inclusão durante as duas etapas de centrifugação (etapas 2.1–2.7 e 1.3.15-1.3.20) é imperativa para inibir as vias de estresse celular, o que resultaria em menores níveis de viabilidade24. Além disso, foi demonstrado que o ROCKi promove o crescimento neurológico e melhora a maturação neuronal25. Assim, a suplementação com ROCKi é continuada por 48 h pós-bioimpressão. No entanto, é imperativo remover a suplementação de ROCKi após 48 h para garantir a lavagem completa durante as mudanças de meios subsequentes antes que as células sejam usadas para ensaio.
Uma etapa adicional que requer atenção crítica é durante a adição de mídia pós-impressão e as alterações de mídia (etapas 2.8-2.13). O arcabouço de hidrogel bioimpresso tem uma rigidez biomecânica equivalente de apenas 1,1 kPa, equivalente à substância cinzenta. Conforme descrito na etapa 2.10, é fundamental pipetar suavemente para o lado do poço durante a adição e aspiração do meio para evitar perturbações. Isso é particularmente relevante para placas de 384 poços, onde o nível de gel ocupa uma proporção maior do volume total do poço. Este método também deve ser usado em poços de controle 2D para evitar o levantamento de borda de células e cisalhamento de excrescências de neuritos. Os autores também gostariam de destacar a importância da técnica estéril dentro da bioimpressora, que deve ser tratada com cautela equivalente à de um gabinete de biossegurança usado para culturas de células derivadas de iPSC. Isso inclui filtragem estéril 70% EtOH e dH2O usados nos procedimentos de greenlighting e impressão, manter tampas nos cartuchos e placas enquanto movem as mãos para dentro e para fora da bioimpressora e descontaminar superfícies dentro da bioimpressora com lenços de etanol 70% antes e depois da impressão.
O arcabouço de hidrogel bioimpresso, formado a partir de soluções de biotinta e ativador, selecionado para desenvolver este modelo é selecionado a partir de uma gama de biotintas e soluções ativadoras desenvolvidas pela Inventia Life Science para uso dentro da bioimpressora RASTRUM. A laminina e o ácido hialurônico foram identificados como moléculas de relevância para a maturação neuronal derivada da iPSC devido ao seu papel na orientação axonal, formação de sinapses e formação da rede perineuronal26,27. Além disso, uma rigidez biomecânica de 1,1 kPa foi selecionada, uma vez que hidrogéis de baixa densidade demonstraram permitir um melhor crescimento de neuritos dos neurônios12. Se forem feitas modificações no protocolo usando neurônios e astrócitos que foram diferenciados internamente ou de um fornecedor comercial diferente, recomenda-se fazer um teste de seleção de matriz para determinar o arcabouço de hidrogel mais suportável15. Além disso, a densidade celular também pode precisar ser otimizada se mudanças nas fontes celulares forem feitas para garantir a viabilidade ideal e evitar a superlotação de hidrogel. Para todas as modificações e solução de problemas relacionados à função da bioimpressora, os autores recomendam entrar em contato com os fabricantes e referenciar os protocolos do fabricante.
O SNC contém uma ampla gama de subtipos neuronais e células gliais, todos os quais existem em diferentes nichos cerebrais e têm papéis específicos contribuindo para a funçãoneural28. Dentro do contexto dessa ampla abrangência, este modelo representa apenas os dois tipos celulares mais abundantes (astrócitos e neurônios glutamatérgicos excitatórios). Importantes tipos celulares como micróglia, oligodendrócitos e células endoteliais formadoras de barreira hematoencefálica são omitidos desse sistema. A inclusão de microglia pode ser relevante no foco em interações neuroimunes, e oligodendrócitos podem ser de interesse em doenças que afetam a mielinização central. Além de seu papel na patologia, células como as células endoteliais formadoras da barreira hematoencefálica excretam enzimas metabolizadoras de fármacos, o que poderia afetar o uso desse modelo para ensaios farmacocinéticos29. Uma outra limitação do modelo pode ser a proporção de astrócitos para neurônios; A proporção de astrócitos/neurônios varia muito entre as regiões cerebrais, com valores sugeridos entre 1:1 e 1:330,31. Este modelo tem uma proporção aproximada de 1:1,5 astrócitos para neurônios; Assim, esse modelo pode não ser relevante para modelar áreas cerebrais onde os astrócitos são mais abundantes, como em áreas da substância branca30.
Outros protocolos para o desenvolvimento de modelos de cocultura bioimpressos 3D têm sido publicados nos últimos anos. Uma publicação de Sullivan et al., 2021, apresentou um modelo neural bioimpresso 3D usando células progenitoras neurais derivadas de iPSC, que demonstra alta viabilidade pós-impressão e aprimoramento da função neuronal em comparação com culturas 2D32. Entretanto, neste protocolo, células progenitoras neurais foram utilizadas como fonte celular e mantidas em cultura por 4 semanas. Neste protocolo, neurônios glutamatérgicos derivados de iPSC e astrócitos comercialmente disponíveis foram usados. Isso permite que uma rede 3D de células co-cultivadas seja estabelecida em apenas 7 dias; Como demonstrado pela análise de crescimento de neuritos, o crescimento de neuritos começa dentro de 24 h e continua de forma linear durante todo o período de 156 h para o qual o crescimento celular foi monitorado. O rápido estabelecimento dessas redes pode ser atribuído, em parte, ao uso de neurônios glutamatérgicos que utilizam a expressão gênica otimizada de NGN2 induzível por doxiciclina, que mostra a expressão de marcadores maduros do subtipo neuronal em 7 dias, mesmo em cultura 2D33. O encurtamento desse período de crescimento com o uso dessa técnica é importante para a implementação de modelos na indústria biofarmacêutica, uma vez que o desenvolvimento de ensaios requer rápido retorno e desenvolvimento de modelos celulares15.
Em conclusão, este modelo mostra potencial para um modelo 3D de neurônios e astrócitos, que é estabelecido de forma rápida e conveniente para fins de triagem. Aplicações futuras para este tipo de modelo poderiam ser para esforços de descoberta de drogas em diferentes doenças do SNC, com a oportunidade de expandir para diferentes doenças usando linhagens iPSC de pacientes ou doenças editadas geneticamente. Além disso, o uso de neurônios glutamatérgicos derivados da expressão de NGN2 induzida por doxiciclina iPSC permite que as células atinjam a maturidade em menos tempo, o que pode ser utilizado para o desenvolvimento de modelos do cérebro envelhecido para pesquisa em neurodegeneração. Este sistema também pode ser expandido através do uso de tipos celulares adicionais em cocultura, incluindo micróglia e oligodendrócitos.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Os autores gostariam de agradecer a Alex Volkerling, Martin Engel e Rachel Bleach por sua assistência no desenvolvimento do protocolo e feedback sobre o manuscrito.
Materials
| 2-mercaptoethanol | Thermofisher | 31350010 | |
| 384-well plate | PerkinElmer | 6057300 | |
| 96-well plate | PerkinElmer | 6055300 | |
| Activator fluid F299 | Inventia Life Science | N/A | |
| Activator fluid F3 | Inventia Life Science | N/A | |
| B27 (50x) minus Vit A | Thermofisher | 12587010 | |
| Bioink fluid F261 | Inventia Life Science | N/A | |
| Bioink fluid F32 | Inventia Life Science | N/A | |
| Doxycycline hyclate | Sigma Aldrich | D5207 | |
| GlutaMAX (100x) | Thermofisher | 35050061 | |
| Goat anti-mouse IgG H&L Alexa Fluor 647 | Abcam | ab150115 | |
| Goat anti-rabbit IgG H&L Alexa Fluor 488 | Abcam | ab150077 | |
| Hoechst | Abcam | ab228551 | |
| Human BDNF Recombinant Protein | Thermofisher | PHC7074 | |
| Human NT3 Recombinant Protein | Thermofisher | PHC7036 | |
| iCell Astrocytes | Fujifilm CDI | 1434 | |
| INCell Analyser 6500HS | Molecular Devices | N/A | high content imaging system |
| Incucyte S3 | Sartorius | N/A | |
| ioGlutamatergic Neurons (Large vial) | Bit.bio | e001 | |
| Laminin (1 mg/mL) | Sigma Aldrich | L2020 | |
| Live/dead kit (Calcein-AM, Ethidium homo-dimer-1) | Invitrogen | L3224 | |
| Mouse anti-BIII tubulin NL637 conjugated | R&D systems | SC024 | |
| Neurobasal media | Thermofisher | 21103049 | |
| Normal Donkey Serum | Abcam | ab7475 | |
| NucBlue Live (Hoechst 33342) | Thermofisher | R37605 | |
| Opti-MEM | Thermofisher | 11058021 | |
| Paraformaldehyde | Sigma Aldrich | P6148 | |
| PEI 50% in H2O | Sigma Aldrich | 181978 | |
| Pierce Borate Buffer 20x | Thermofisher | 28341 | |
| Prism | GraphPad | Data analysis software | |
| Rabbit anti-ionotropic glutamatre receptor 2 (GluR2) | Abcam | ab206293 | |
| RASTRUM(TM) Bioprinter | Inventia Life Science | N/A | Bioprinter |
| RASTRUM(TM) Bioprinter Cartridges | Inventia Life Science | N/A | Bioprinter Cartridges |
| RASTRUM(TM) Targeting plate | Inventia Life Science | N/A | Targeting plate |
| Rho kinase (ROCK) inhibitor | Abcam | ab120129 | |
| Sheep anti-GFAP NL493 conjugated | R&D systems | SC024 | |
| Signals Image Artist | PerkinElmer | N/A | Image analysis platform |
| Triton X-100 | Thermofisher | HFH10 | |
| Zeiss Axio Observer | Zeiss | N/A | Inverted microscope platform |
References
- Jung, Y. L., Hwang, J., Yoo, H. S. Disease burden metrics the innovations of leading pharmaceutical companies: a global and regional comparative study. Globalization and Health. 16 (1), 80-80 (2020).
- Potkin, S. G., et al. The neurobiology of treatment-resistant schizophrenia: paths to antipsychotic resistance and a roadmap for future research. npj Schizophrenia. 6, 1 (2020).
- Zahra, W., et al., Keswani, C., et al. The Global Economic Impact of Neurodegenerative Diseases: Opportunities and Challenges. Bioeconomy for Sustainable Development. , (2019).
- Perucca, E. The pharmacological treatment of epilepsy: recent advances and future perspectives. Acta Epileptologica. 3 (1), 22 (2021).
- Nikolakopoulou, P., et al. Recent progress in translational engineered in vitro models of the central nervous system. Brain. 143 (11), 3181-3213 (2020).
- Whitehouse, C., Corbett, N., Brownlees, J. 3D models of neurodegeneration: implementation in drug discovery. Trends in Pharmacological Sciences. 44 (4), 208-221 (2023).
- Rauti, R., Renous, N., Maoz, B. M. Mimicking the brain extracellular matrix in vitro: A review of current methodologies and challenges. Israel Journal of Chemistry. 60 (12), 1141-1151 (2020).
- Fawcett, J. W., Oohashi, T., Pizzorusso, T. The roles of perineuronal nets and the perinodal extracellular matrix in neuronal function. Nature Reviews Neuroscience. 20 (8), 451-465 (2019).
- Lam, D., et al. Tissue-specific extracellular matrix accelerates the formation of neural networks and communities in a neuron-glia co-culture on a multi-electrode array. Scientific Reports. 9, 4159 (2019).
- Roll, L., Lessmann, K., Brüstle, O., Faissner, A. Cerebral organoids maintain the expression of neural stem cell-associated glycoepitopes and extracellular matrix. Cells. 11 (5), 760 (2022).
- Yan, Y., Bejoy, J., Marzano, M., Li, Y. The use of pluripotent stem cell-derived organoids to study extracellular matrix development during neural degeneration. Cells. 8 (3), 242 (2019).
- Ma, L., et al. 3D bioprinted hyaluronic acid-based cell-laden scaffold for brain microenvironment simulation. Bio-Design and Manufacturing. 3 (3), 164-174 (2020).
- Liaw, C. -. Y., Ji, S., Guvendiren, M. Engineering 3D hydrogels for personalized in vitro human tissue models. Advanced Healthcare Materials. 7 (4), 1701165 (2018).
- Ma, J., Huang, C. Composition and mechanism of three-dimensional hydrogel system in regulating stem cell fate. Tissue Engineering Part B: Reviews. 26 (6), 498-518 (2020).
- Belfiore, L., et al. Generation and analysis of 3D cell culture models for drug discovery. European Journal of Pharmaceutical Sciences. 163, 105876 (2021).
- Langhans, S. A. Three-dimensional in vitro cell culture models in drug discovery and drug repositioning. Frontiers in Pharmacology. 9, 6 (2018).
- Engel, M., Belfiore, L., Aghaei, B., Sutija, M. Enabling high throughput drug discovery in 3D cell cultures through a novel bioprinting workflow. SLAS Technology. 27 (1), 32-38 (2022).
- Takamura, T., et al. Influence of age on global and regional brain stiffness in young and middle-aged adults. Journal of Magnetic Resonance Imaging. 51 (3), 727-733 (2020).
- Slanzi, A., Iannoto, G., Rossi, B., Zenaro, E., Constantin, G. In vitro models of neurodegenerative diseases. Frontiers in Cell and Developmental Biology. 8, 328 (2020).
- de Souza, N. Organoid variability examined. Nature Methods. 14 (7), 655-655 (2017).
- Hernández, D., et al. Culture variabilities of human iPSC-derived cerebral organoids are a major issue for the modelling of phenotypes observed in Alzheimer’s disease. Stem Cell Review and Reports. 18 (2), 718-731 (2022).
- Li, R., et al. Differentiation of human iPS cells into sensory neurons exhibits developmental stage-specific cryopreservation challenges. Frontiers in Cell and Developmental Biology. 9, 796960 (2021).
- Nishiyama, Y., et al. Safe and efficient method for cryopreservation of human induced pluripotent stem cell-derived neural stem and progenitor cells by a programmed freezer with a magnetic field. Neuroscience Research. 107, 20-29 (2016).
- Uhrig, M., Ezquer, F., Ezquer, M. Improving cell recovery: Freezing and thawing optimization of induced pluripotent stem cells. Cells. 11 (5), 799 (2022).
- Harbom, L. J., et al. The effect of rho kinase inhibition on morphological and electrophysiological maturity in iPSC-derived neurons. Cell and Tissue Research. 375 (3), 641-654 (2019).
- Koh, H. S., Yong, T., Chan, C. K., Ramakrishna, S. Enhancement of neurite outgrowth using nano-structured scaffolds coupled with laminin. Biomaterials. 29 (26), 3574-3582 (2008).
- Tarus, D., et al. Design of hyaluronic acid hydrogels to promote neurite outgrowth in three dimensions. ACS Applied Materials & Interfaces. 8 (38), 25051-25059 (2016).
- Brain Initiative Cell Census Network (BICCN). Initiative Cell Census Network (BICCN). A multimodal cell census and atlas of the mammalian primary motor cortex. Nature. 598 (7879), 86-102 (2021).
- Dauchy, S., et al. Expression and transcriptional regulation of ABC transporters and cytochromes P450 in hCMEC/D3 human cerebral microvascular endothelial cells. Biochemical Pharmacology. 77 (5), 897-909 (2009).
- Herculano-Houzel, S. The glia/neuron ratio: How it varies uniformly across brain structures and species and what that means for brain physiology and evolution. Glia. 62 (9), 1377-1391 (2014).
- von Bartheld, C. S., Bahney, J., Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. The Journal of Comparative Neurology. 524 (18), 3865-3895 (2016).
- Sullivan, M. A., et al. 3D bioprinting of stem cell-derived central nervous system cells enables astrocyte growth, vasculogenesis and enhances neural differentiation/function. bioRxiv. , (2022).
- Pawlowski, M., et al. Inducible and deterministic forward programming of human pluripotent stem cells into neurons, skeletal myocytes, and oligodendrocytes. Stem Cell Reports. 8 (4), 803-812 (2017).
