1. Isolation and fixation of inner ears
- For analysis of cellular patterning in the organ of Corti, euthanize appropriately staged embryos or adults.
- Dissect out the otic capsule, containing the membranous inner ear. In the mouse, this can be easily done on embryos at embryonic (E) day 14.5 and older, in which E0.5 is the day at which a vaginal plug was detected. Dissection of the otic capsule is accomplished first by decapitation and opening of the skull at the midline. Remove brain to expose each ear (Fig. 1A). Inner ears can be shelled out intact using forceps (such as Dumont #5, Fig. 1B). If imaging the organ of Corti, careful removal of all neuronal and connective tissue associated with the otic capsule is not necessary.
- Fix whole inner ears by immersion in 4% paraformaldehyde (PFA) with gentle rocking at 4°C overnight in 1.8 ml screw-capped NUNC Cryo-tube vials, or other appropriately sized container. PFA solution should be made fresh in ddH20, then stored at -20°C until needed.
- The next day, wash out the fixative with three changes of 1X phosphate buffered saline (PBS). Tissue can be stored in sterile 1X PBS for days up to months until ready for processing. For adult tissue, decalcify in 10% EDTA (0.27 M) at 4°C for 5 days, followed by rinsing and storage in 1X PBS.
2. Cutting vibratome sections
- Prepare 4% Low Melting Point agarose in 1X PBS. Add low melting point agarose to 1X PBS and microwave until agarose is in solution. Take care that the solution does not bubble over – take out of the microwave periodically and swirl to mix. Keep solution in a 55°C water bath until ready to use.
- Using forceps, remove inner ear from 1X PBS solution and position at the bottom of a Peel-A-Way embedding mold. Pipet away any excess 1X PBS from the mold. Fill mold to cover the tissue with plenty of liquid 4% Low Melting Point agarose (Fig. 2A).
- Position the inner ear for sectioning in the appropriate plane. I have obtained good cross-sections of the organ of Corti by approximating the position of the otic capsule as if it had been left in situ in the head and sectioning in the transverse plane. For E14.5 embryos to adults, position the inner ear so that it is viewed laterally, with the lateral semi-circular canal visible (Fig. 2B). Angle the inner ear so that the line formed by the posterior side of the otic capsule is approximately at a 45-degree angle from the intended plane of sectioning (Fig. 2A, B). When the otic capsule is viewed so that the dorsal portion of the otic capsule is at the top, the basal turn of the cochlea should be located at the bottom (arrowhead, Fig. 2B) and the anterior semi-circular canal (asc) should be at the top (Fig. 2B).
- Agarose will set at RT within 30 mins. Transfer Peel-Away molds containing embedded tissue to 4°C until ready to section. If sectioning will be put off until the next day, add 1X PBS to cover the agarose in the mold and store in a box containing moist paper towels soaked in 1X PBS.
- Peel away the plastic mold. Using a razor blade, cut out an agarose box containing the inner ear tissue. Take care to ensure that the plane of section is straight and parallel to the opposite side of the block, which will be the surface on which the agarose block is glued down. Since the agarose does not infiltrate the tissue, do not trim too much agarose from around the tissue, so that the tissue will be well-supported.
- Use superglue to attach the block to the cutting surface.
- Section at 40 to 100 μm thickness by vibratome. Save sections in a tissue-culture dish filled with 1X PBS, 0.1% TritonX-100. A representative section through the cochlear spiral is shown in Fig. 2C.
- Use forceps to dissect away the agarose from the tissue. Agarose will not penetrate the tissue, but will just be stuck to the outer surface of the inner ear.
- If desired, pool agarose-free, sectioned tissue using a pipet with a large-hole opening to prevent excessive shearing of the tissue. If performing multiple antibody stains on multiple different tissues, sections can be organized in a 16- or 24-well tissue culture dish.
3. Antibody staining vibratome sections
Proceed with antibody stain. Process sections in whole-mount, free-floating in solution. Below is an example antibody staining protocol for the S100 marker of the pillar cells of the organ of Corti, but the protocol can be used for any antibody. All washes are conducted at room temperature unless otherwise noted.
- In a multi-well tissue culture dish, incubate free-floating sections with gentle rocking in PBT (1X PBS, 1% bovine serum albumin, 0.1% Triton X-100) for 30 mins.
- Pipet off old solution and block free-floating sections with gentle rocking in PBT + normal goat serum (50 μl NGS/ 1 ml PBT) for 30 mins.
- Pipet off old solution and add primary antibody diluted in PBT + NGS (1:200 dilution of S100 in PBT+NGS). Incubate overnight at 4°C with gentle rocking.
- Pipet off primary antibody solution. Wash 3X, 5 mins. with PBT, then 4X, 30 mins. with PBT.
- Block with PBT + NGS, 30 mins.
- Incubate with fluorescently-conjugated secondary antibody (raised in goat) diluted in PBT + NGS. Incubate overnight at 4°C, or a few hours, RT, gentle rocking.
- Pipet off secondary antibody solution. Wash 3X, 5 mins., then 4X, 30 mins. with PBT.
- Pipet off last PBT wash and replace with appropriate aqueous mounting medium containing a nuclear counterstain.
4. Mounting sections on slides and confocal microscopy
To prevent crushing the thick sections with the coverslip, when mounting individual sections, a spacer must be inserted between the microscope slide and the coverslip. Below are two alternative methods of mounting thick vibratome sections:
-
- A spacer can be created by placing a rim of vacuum grease in a square around the tissue section. Thus, when laying down the coverslip, the edges of the coverslip will be sealed with vacuum grease. Press down on the edges of the coverslip with your thumb to firmly adhere it onto the slide. If the vacuum grease is neatly and uniformly present around all edges of the coverslip, the slide will be sealed sufficiently for use on an inverted confocal microscope.
- Alternatively, a spacer can be created using agarose chromatography resin of known mesh size (such as Affi-Gel Blue Gel, Bio-Rad). Pipet 1 μl of resin onto the microscope slide in each of the four corners where the coverslip will be placed. Pipet one tissue section and mounting media into the middle of the four dots of resin. Coverslip. If using an inverted confocal microscope, seal the coverslip onto the microscope slide using nailpolish.
- Obtain optical sections of the stained vibratome slices using confocal microscopy. To quantify various cell types within the organ of Corti, obtain a z-stack at a defined step size (between 1.5 and 3 μm). Sections should be counter-stained with a nuclear dye (step 3.8). Images can be examined on any computer screen after confocal image collection. Cells can be counted manually be observing the appearance and disappearance of nuclei as images are examined sequentially through the z-stack.
5. Representative Results:
A representative confocal z-stack through a vibratome section, stained for S100 protein, which is present in pillar and Deiters’ cells, and counterstained with a nuclear dye (Fig. 3A-F). Each panel is the confocal image taken at 3 μm-steps, with panel A the confocal image obtained closest to the surface of the vibratome section and panel F the confocal image obtained most internally within the vibratome section. The morphology of the organ of Corti appears minimally disrupted. In particular, the cytoplasmic extensions of the pillar cells are not broken. By noting the appearance and disappearance of pillar cell nuclei through the z-stack (Fig. 3A to 3F), both inner and outer pillar cell number can be counted (see numbering, Fig. 3A – F). Due to the small number of each cell type per confocal image, this approach can be used to count most cell types within the organ of Corti.
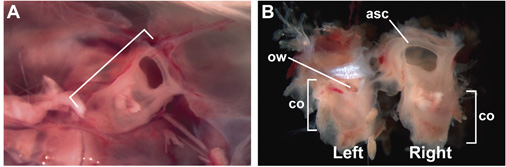
Figure 1. Intact, 15-week-old, adult inner ears encased in the otic capsule. (A) The right otic capsule (bracketed) encased in the skull, viewed from a vantage point of inside the head, after removal of the brain. Anterior is to the right. (B) Intact, 15-week-old inner ears after dissection from the skull. Lateral surface of the left inner ear is visible; medial surface of the right inner ear is visible. Labels indicate the locations of the anterior semi-circular canal (asc), co (cochlea), and ow (oval window).
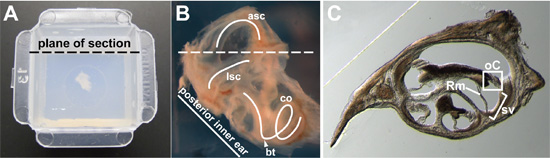
Figure 2. (A) Whole inner ear from a 15-week-old adult, embedded in agarose in the center of a Peel-Away mold. Intended plane of section is indicated. (B) Close-up image of a 15-week-old, whole inner ear prior to embedding. A portion of the surrounding bone has been dissected away so the membranous labyrinth could a viewed more clearly. The lateral semi-circular canal (lsc), anterior semi-circular canal (asc), and cochlea (co) are traced (white curves). The basal turn of the cochlea (arrowhead) and intended plane of section (dashed line) are indicated. (C) Representative cross-section through the cochlea. In one cross-section through the cochlear duct, the organ of Corti (oC) is boxed, the stria vascularis (sv) is bracketed, and the Reissner’s membrane (Rm) is indicated.
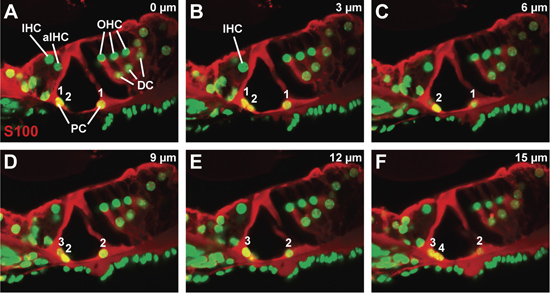
Figure 3. The organ of Corti in a wild-type mouse at P21. (A-F) Sequential confocal series, at a 3 μm step-size, through a S100 antibody-stained organ of Corti to visualize the pillar cells and Deiters’ cells. Counts of inner and outer pillar cells from the start to the end of the series are numbered. Abbreviations: IHC (inner hair cell), aIHC (adjacent inner hair cell), OHC (outer hair cell), PC (pillar cell), DC (Deiters’ cell).
