In de afgelopen twee decennia hebben vaste stoffen en films die zijn gemaakt van geaggregeerde organische moleculen meerdere toepassingen gevonden in opto-elektronische apparaten. Apparaten op basis van dergelijke materialen hebben veel aantrekkelijke eigenschappen, waaronder een klein gewicht, flexibiliteit, een laag energieverbruik en potentieel voor goedkope productie met inkjetafdrukken. Displays op basis van organische lichtgevende diodes (OLED’s) vervangen vloeibare kristallijne displays als state of the art voor mobiele telefoons, laptops, televisietoestellen en andere elektronische apparaten1,,2,3,4. Het belang van OLED’s voor verlichtingstoepassingen zal naar verwachting toenemen in de komende jaren4. De prestaties van organische fotovoltaïsche apparaten is gestaag verbeteren, met energie conversie efficiëntie van meer dan 16% onlangs gemeld voor single-junction organische zonnecellen5. Organische materialen hebben ook het potentieel om andere technologieën te verstoren, zoals glasvezelcommunicatie, waarbij het gebruik ervan de ontwikkeling van elektro-optische modulatoren met extreem hoge bandbreedtes van 15 THz en bovende 6,7mogelijkmaakt .
Een grote uitdaging bij het optimaliseren van solid-state moleculaire materialen voor toepassingen in opto-elektronica is dat hun eigenschappen meestal sterk afhangen van de nanoschaalstructuur van het materiaal. Het productieproces maakt het mogelijk om de nanostructuur van een materiaal tot op zekere hoogte te definiëren met behulp van gecontroleerde groeitechnieken, zoals chemische dampafzetting,8 templating van optisch actieve moleculen op een ander materiaal (d.w.z. een polymeermatrix9,10),thermische annealing11,12,enz. Echter, nanoschaal stoornis is intrinsiek aan de meeste moleculaire materialen en meestal niet volledig kan worden geëlimineerd. Daarom is begrijpen hoe wanorde de eigenschappen van een materiaal beïnvloedt en manieren vinden om het te ontwerpen voor optimale prestaties essentieel voor het rationele ontwerp van organische opto-elektronische materialen.
De mate van wanorde in moleculaire materialen is gewoonlijk te groot om het als verstoring van een periodieke kristallijne structuur met een elektronische structuur te behandelen die door bandtheorie kan worden beschreven. Aan de andere kant is het aantal moleculen dat in een simulatie moet worden opgenomen om de eigenschappen van een bulkmateriaal of een film te reproduceren te groot om eerste principes te gebruiken voor kwantumchemische methoden zoals dichtheidsfunctionele theorie (DFT)13,,14 en tijdafhankelijke dichtheidsfunctionele theorie (TD-DFT)15,16. Organische moleculen met toepassingen in opto-elektronica hebben doorgaans relatief grote π-geconjugeerde systemen; velen hebben ook donor- en acceptorgroepen. Het vastleggen van de juiste lading-overdracht gedrag in dergelijke moleculen is essentieel voor het berekenen van hun opto-elektronische eigenschappen, maar het kan alleen worden bereikt met behulp van lange afstand gecorrigeerde hybride functionals in TD-DFT17,18,19,20. Berekeningen die gebruik maken van dergelijke functionalen schaal super lineair met de grootte van het systeem en, op dit moment, ze zijn alleen praktisch voor het modelleren van de opto-elektronische eigenschappen van individuele organische moleculen of kleine moleculaire aggregaten die kunnen worden beschreven met behulp van niet meer dan ~ 104 atomaire basisfuncties. Een simulatiemethode die wanordelijke materialen zou kunnen beschrijven die uit grote aantallen chromophores bestaan zou zeer nuttig zijn voor het modelleren van deze systemen.
De omvang van intermoleculaire interacties in moleculaire materialen is vaak vergelijkbaar met of kleiner dan de volgorde van variatie in de energetische parameters (zoals de eigenstate energieën of excitatie-energieën) tussen individuele moleculen waaruit het materiaal bestaat. In dergelijke gevallen is multiscale modellering de meest veelbelovende benadering van het begrijpen en optimaliseren van de opto-elektronische eigenschappen van grote wanordelijke moleculaire systemen21,22,23. Deze aanpak maakt gebruik van eerste principes kwantumchemische methoden (meestal DFT en TD-DFT) om nauwkeurig te berekenen van de eigenschappen van individuele moleculen die het materiaal samenstellen. De Hamiltonian van een materiaalmonster dat groot genoeg is om het bulkmoleculaire materiaal te vertegenwoordigen (misschien, door gebruik te maken van periodieke randvoorwaarden) wordt vervolgens geconstrueerd met behulp van de parameters die werden berekend voor individuele moleculen. Deze Hamiltoniaan kan vervolgens worden gebruikt om de opto-elektronische parameters van een groot moleculair aggregaat, een dunne film of een bulk moleculair materiaal te berekenen.
Exciton modellen zijn een subklasse van multiscale modellen waarin opgewonden toestanden van een moleculair materiaal worden vertegenwoordigd in een basis van excitons: elektronen-gat paren die gebonden zijn door Coulomb attractie24,25. Voor het modelleren van veel opgewonden staatprocessen, is het voldoende om alleen Frenkel excitons26, waar het elektron en het gat zijn gelokaliseerd op hetzelfde molecuul. Ladingoverdrachtexcitons, waarbij het elektron en het gat op verschillende moleculen zijn gelokaliseerd, moeten mogelijk in sommige gevallen worden opgenomen (bijvoorbeeld bij het modelleren van ladingsscheiding in donoracceptorsystemen)27,28. Hoewel exciton modellen zijn multiscale modellen die kunnen worden geparameterd met behulp van alleen eerste-principe berekeningen op individuele moleculen, ze nog steeds goed voor intermoleculaire interacties. De twee primaire interactietypen die zij kunnen verantwoorden zijn (a) excitoonse koppelingen tussen moleculen die het vermogen van excitons karakteriseren om over moleculen heen te delokaliseren of over te brengen en (b) elektrostatische polarisatie van de elektronendichtheid op een molecuul door de ladingsverdeling op omringende moleculen. We hebben eerder aangetoond dat beide factoren belangrijk zijn voor het modelleren van de optische en elektro-optische eigenschappen van moleculaire aggregaten, zoals de optische absorptiespectra29 en de eerste hyperpolarisatatoren30.
In dit artikel presenteren we een protocol voor het parametriseren van excitonmodellen die kunnen worden gebruikt om de optische spectra en andere opto-elektronische eigenschappen van grote moleculaire aggregaten en bulkmoleculaire materialen te berekenen. De excitonische Hamiltonian wordt verondersteld om een strak-bindende Hamiltonian24,25te zijn,
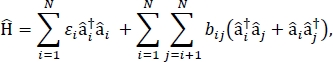
waar εi de excitatie-energie van het ith-molecuul in het materiaal is, is bij de excitoonse koppeling tussen de ith- en de jth-moleculen, â i† en â i zijn respectievelijk de creatie- en vernietigingsoperatoren voor een opgewonden toestand op het ith-molecuul in het materiaal. De excitonische Hamiltonian parameters worden gevonden met behulp van TD-DFT berekeningen die worden uitgevoerd op individuele moleculen die deel uitmaken van het materiaal. In deze TD-DFT berekeningen, de lading verdeling op alle andere moleculen in het materiaal wordt vertegenwoordigd door elektrostatische inbedding van atoompunt ladingen om rekening te houden met elektrostatische polarisatie van de elektronische dichtheid van een molecuul. De excitatie-energieën, εi, voor individuele moleculen worden rechtstreeks uit de TD-DFT-berekeningsoutput gehaald. De excitonische koppelingen, bij, tussen moleculen worden berekend met behulp van de overgangsdichtheidkubusmethode31, met de grond-naar-opgewonden toestandsovergangsdichtheid voor de interagerende moleculen uit de output van een TD-DFT-berekening in Gaussian32 en post-verwerkt met behulp van de Multiwfn multifunctionele golfanalyzfunctioner33. Voor het simuleren van de eigenschappen van bulkmoleculaire vaste stoffen kunnen periodieke randvoorwaarden worden toegepast op de Hamiltonian.
Het huidige protocol vereist dat de gebruiker toegang heeft tot de Gaussian32- en Multiwfn33-programma’s. Het protocol is getest met Gaussian 16, revisie B1 en Multiwfn versie 3.3.8, maar moet ook werken voor andere recente versies van deze programma’s. Daarnaast maakt het protocol gebruik van een aangepaste C++ utility en een aantal aangepaste python 2.7 en Bash scripts, waarvan de broncode wordt geleverd onder de GNU General Public License (Versie 3) op https://github.com/kocherzhenko/ExcitonicHamiltonian. De berekeningen zijn bedoeld om te worden uitgevoerd op een machine met een besturingssysteem uit de Unix/Linux-familie.
