Son yirmi yılda, birleştirilmiş organik moleküllerden yapılan katı maddeler ve filmler optoelektronik cihazlarda birden fazla uygulama bulmuştür. Bu tür malzemelere dayalı cihazlar, küçük ağırlık, esneklik, düşük güç tüketimi ve mürekkep püskürtmeli baskı kullanarak ucuz üretim potansiyeli gibi birçok çekici özelliğe sahiptir. Organik ışık yayan diyotlar dayalı görüntüler (LED’ler) cep telefonları, dizüstü bilgisayarlar, televizyon setleri ve diğer elektronikcihazlariçin sanat durumu olarak sıvı kristal ekranlar yerine 1,2,3,4. Aydınlatma uygulamaları için LED’lerin öneminin önümüzdeki yıllarda artması beklenmektedir4. Organik fotovoltaik cihazların performansı sürekli olarak iyileşiyor, güç dönüşüm verimliliği üzerinde 16% son zamanlarda tek kavşakorganik güneş pilleri için rapor5. Organik maddeler de diğer teknolojileri bozmak için potansiyele sahip, fiber optik iletişim gibi, bunların kullanımı 15 THz ve üzerinde son derece yüksek bant genişlikleri ile elektro-optik modülatörlerin geliştirilmesini sağlarnerede 6,7.
Optoelektronik uygulamaları için katı hal moleküler malzemelerin optimize önemli bir sorun genellikle özellikleri kuvvetle malzemenin nanoölçekli yapısına bağlıdır. Üretim süreci, kimyasal buharbirikimi, optik olarak aktif moleküllerin başka bir malzemeye (yani polimer matris9,10), termal annealing11,12, vb. üzerine 8 cezbedici olarak kontrollü büyüme teknikleri kullanılarak bir malzemenin nanoyapısını tanımlamaya olanak sağlar. Ancak, nanoölçekli bozukluk en moleküler malzemeler için içsel ve genellikle tamamen ortadan kaldırılamaz. Bu nedenle, bozukluğun bir malzemenin özelliklerini nasıl etkilediğini anlamak ve onu optimum performans için tasarlamanın yollarını bulmak organik optoelektronik malzemelerin rasyonel tasarımı için gereklidir.
Moleküler malzemelerdeki bozukluk derecesi genellikle bant teorisi ile tarif edilebilen elektronik bir yapıile periyodik kristal yapının bir pertürbasyon olarak tedavi etmek için çok büyük. Öte yandan, bir toplu malzeme veya bir film özelliklerini çoğaltmak için bir simülasyon dahil edilmesi gereken moleküllerin sayısı yoğunluk fonksiyonel teorisi (DFT)13,14 ve zamana bağlı yoğunluk fonksiyonel teorisi (TD-DFT)15,16gibi ilk ilkeleri kuantum kimyasal yöntemleri kullanmak için çok büyük . Optoelektronik uygulamaları ile organik moleküller genellikle nispeten büyük π-konjuge sistemlere sahip; birçok da donör ve kabul grupları var. Bu tür moleküllerde doğru şarj-transfer davranışı yakalama onların optoelektronik özellikleri hesaplamak için gereklidir, ama sadece TD-DFT17,18,,19,20uzun menzilli düzeltilmiş hibrid fonksiyoneller kullanılarak gerçekleştirilebilir . Bu tür işlevleri kullanan hesaplamalar sistemin büyüklüğü ile süper doğrusal ölçeklenir ve şu anda sadece bireysel organik moleküllerin veya ~104 atomik temel işlevlerinden fazla olmayan küçük moleküler agregaların optoelektronik özelliklerini modellemek için pratiktir. Çok sayıda kromofordan oluşan düzensiz materyalleri tanımlayabilen bir simülasyon yöntemi bu sistemlerin modelalınmasında çok yararlı olacaktır.
Moleküler malzemelerdeki moleküller arası etkileşimlerin büyüklüğü genellikle malzemeyi oluşturan tek tek moleküller arasındaki enerjik parametrelerdeki (eigenstate enerjileri veya uyarma enerjileri gibi) değişim sırası ile karşılaştırılabilir veya daha küçüktür. Bu gibi durumlarda, çok ölçekli modelleme anlamak ve büyük düzensiz moleküler sistemlerin optoelektronik özellikleri optimize etmek için en umut verici bir yaklaşımdır21,22,23. Bu yaklaşım, malzemeyi oluşturan tek tek moleküllerin özelliklerini doğru bir şekilde hesaplamak için ilk prensipleri kuantum kimyasal yöntemlerini (genellikle DFT ve TD-DFT) kullanır. Toplu moleküler malzemeyi temsil edecek kadar büyük bir malzeme örneğinin Hamiltonian ‘ı (belki de periyodik sınır koşulları kullanılarak) tek tek moleküller için hesaplanan parametreler kullanılarak oluşturulur. Bu Hamiltonian daha sonra büyük bir moleküler agrega, ince bir film veya toplu moleküler malzeme nin optoelektronik parametrelerini hesaplamak için kullanılabilir.
Exciton modelleri, moleküler bir maddenin heyecanlı durumlarının eksitonlartemelinde temsil edildiği çok ölçekli modellerin bir alt sınıfıdır : Coulomb cazibe24,25ile bağlı elektron deliği çiftleri . Birçok heyecanlı durum süreçlerini modellemek için, sadece Frenkel eksitonları dahil etmek yeterlidir26, elektron ve delik aynı molekül üzerinde lokalize nerede. Elektron ve deliğin farklı moleküller üzerinde lokalize olduğu yük transferi eksitonları bazı durumlarda (örneğin, donör-kabul edici sistemlerde şarj ayrımı modellenirken)27,28. Exciton modelleri, tek tek moleküller üzerinde sadece ilk prensip hesaplamaları kullanılarak parametrize edilebilen çok ölçekli modeller olmasına rağmen, yine de moleküller arası etkileşimleri hesaba katmaktadır. (a) eksitonların moleküller arasında yerelleşme veya moleküller arasında aktarım yeteneğini karakterize eden moleküller arasındaki iki birincil etkileşim türü ve (b) etrafındaki moleküller üzerindeki yük dağılımı ile bir molekülüzerindeki elektron yoğunluğunun elektrostatik polarizasyonudur. Daha önce bu faktörlerin her ikisi nin de optik soğurmaspektrum29 ve ilk hiperpolarizabilities30gibi moleküler agregaların optik ve elektro-optik özelliklerini modellemek için önemli olduğunu göstermiştir.
Bu yazıda, büyük moleküler agregaların ve dökme moleküler malzemelerin optik spektrumlarını ve diğer optoelektronik özelliklerini hesaplamak için kullanılabilecek exciton modellerinin parametrizeedilmesi için bir protokol sunacağız. Eksitonik Hamiltonian sıkı bağlayıcı Hamiltonian24,,25olduğu varsayılır,
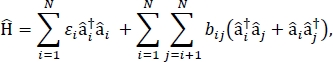
nerede εi malzemedeki ith molekülün uyarma enerjisi, bij ive jth molekülleri arasında eksitonik bağlantı, â i† ve â i yaratılış ve imha operatörleri, sırasıyla, malzemede ith molekülüzerinde heyecanlı bir durum için. Eksitonik Hamilton parametreleri, malzemeyi oluşturan tek tek moleküller üzerinde yapılan TD-DFT hesaplamaları kullanılarak bulunur. Bu TD-DFT hesaplamalarında, malzemedeki diğer tüm moleküllerin yük dağılımı, bir molekülün elektronik yoğunluğunun elektrostatik polarizasyonu için atomik nokta yüklerinin elektrostatik katıştırılması ile temsil edilir. Uyarma enerjileri, εi, bireysel moleküller için doğrudan TD-DFT hesaplama çıkışından alınır. Eksitonik kaplinler, bij, moleküller arasında geçiş yoğunluğu küp yöntemi kullanılarak hesaplanır31, Gaussianbir TD-DFT hesaplama çıkışı alınan etkileşim molekülleri için zemin-heyecanlı devlet geçiş yoğunlukları ile32 ve post-multiwfn çok fonksiyonlu dalga fonksiyonu analizörü kullanılarak işlenir33. Multiwfn Dökme moleküler katıların özelliklerini simüle etmek için Hamiltonian’a periyodik sınır koşulları uygulanabilir.
Geçerli protokol, kullanıcının Gaussian32 ve Multiwfn33 programlarına erişebulaşmasını gerektirir. Protokol Gaussian 16, revizyon B1 ve Multiwfn sürüm 3.3.8 kullanılarak test edilmiştir, ancak bu programların diğer son sürümleri için de çalışması gerekir. Buna ek olarak, protokol özel bir C++ yardımcı programı ve özel python 2.7 ve Bash komut dosyaları, https://github.com/kocherzhenko/ExcitonicHamiltonian GNU Genel Kamu Lisansı (Sürüm 3) altında sağlanan kaynak kodu bir dizi kullanır. Hesaplamalar Unix/Linux ailesinden bir işletim sistemi çalıştıran bir makinede yapılması amaçlanmıştır.
