Figure 4 summarizes the expected SDS depletion following vial-based or cartridge-facilitated precipitation of proteins in a disposable filter cartridge using acetone. Conventional overnight incubation (-20 °C) in acetone is compared to the rapid acetone precipitation protocol at room temperature (step 2), as well as CMW precipitation (step 4). Residual SDS was quantified by the methylene blue active substances (MBAS) assay29. Briefly, 100 µL sample was combined with 100 µL MBAS reagent (250 mg methylene blue, 50 g sodium sulfate, 10 mL sulfuric acid, diluted in water to 1.0 L), followed by the addition of 400 µL chloroform and absorbance measurement of the organic layer at 651 nm on a UV/Vis spectrophotometer. All approaches reduce SDS to permit optimal MS analysis.
Quantitative and reproducible protein recovery is achieved following rapid acetone precipitation and CMW precipitation, as seen in Figure 5 through SDS PAGE analysis of a processed yeast total cell lysate. Precipitation in a disposable filtration cartridge eliminates the need to carefully pipet the SDS-containing supernatant while retaining the aggregated proteins above a membrane filter. Consistent recovery is obtained with all precipitation protocols, with no visible bands detected in the supernatant fractions across three independent replicates.
Figure 6 quantifies the expected yields, including the resolubilization of precipitated protein pellets using cold formic acid (step 6). CMW precipitation affords quantitative recovery by carefully preserving the pellet in a vial-based approach (step 5), which equals that obtained using the cartridge (100 ± 4% vs. 101 ± 3%, respectively). Recovery of acetone-precipitated protein pellets benefits from a filtration cartridge, with a 15-20 % improvement in yield observed. In vials, isolation of the acetone supernatant from the aggregated protein essentially relies on the adherence of the pellet to the PP tube surface; the filtration cartridge eliminates this concern as the filter ensures high recovery of precipitated protein without pipetting.
To efficiently recover LMW proteins and peptides, the acetone precipitation protocol is modified by substituting NaCl for ZnSO4 and raising the solvent percentage to 97%. Combining this specific salt and higher levels of organic solvent are required for the high recovery of LMW proteins and peptides26. As seen in Figure 7, cartridge-based protein precipitation demonstrates superior recovery of a pepsin-digested sample of bovine plasma relative to vial-based precipitation. The disposable spin cartridge can recover over 90% of LMW peptides. More significant differences in yield are noted in the cartridge when employing NaCl, confirming the importance of salt type to maximize yield. Including ZnSO4 as opposed to NaCl results in an aggregated protein pellet that is more readily trapped by the spin cartridge filter.
To assess the efficacy of precipitating proteins over a wide dynamic range, a mixture of three standard proteins was processed: β-galactosidase (β-gal) from E. coli, cytochrome c (Cyt c) from bovine, and enolase (Eno) from S. cerevisiae. The mass ratio of β-gal:Cyt c:Eno was 10,000:10:1. Samples initially contained 2% of SDS prior to cartridge-based precipitation (step 5) and were re-solubilized and digested with trypsin (steps 6 and 7). Samples prepared in vials acted as a control, having no SDS and omitting the precipitation. All samples were subject to equivalent SPE clean-up (step 8). Bottom-up MS was conducted, with MS/MS spectra searched against a combined database containing all proteins from the three species involved (see Table of Materials for instrument and software platforms). A peptide false discovery rate of 1% was employed. All three proteins were identified by MS, with 666, 28, and 35 unique peptides for β-gal, Cyt c, and Eno, respectively. Figure 8 quantifies the relative ratio (peptide peak intensity) from each sample, with a ratio above 1 reflecting a higher peptide abundance for samples processed in the disposable filter cartridges. The results demonstrate the benefits of incorporating SDS into a proteomics workflow, minimizing protein loss (e.g., from potential adsorption to the sample vial), and maximizing peptide yields.
Bovine liver was procured at a local grocery store. The proteins were isolated by extracting the tissue with a solution of 1% SDS. Subsequently, the recovered proteome was precipitated, re-solubilized (urea), and digested with trypsin, all within a disposable cartridge. Bottom-up LC-MS/MS was conducted, resulting in the identification of an average of ~8,000 proteins (~30,000 peptides). False discovery rates of 0.5% and 1.0% for peptide spectra and protein groups, was employed, searching the bovine database. The technical reproducibility of this cartridge-based workflow is assessed through overlapping protein identifications. The replicate MS injections of a common digested sample achieves on average 78 ± 0.5% overlap with the identified proteins. By comparison, samples independently prepared in discrete cartridges achieved 76 ± 0.5% overlap. These data suggest that the contribution of sample preparation toward the total variability of the analysis is minor, relative to that already contributed by the LC-MS instrumental approach. The bovine proteins identified from three technical replicates (processed independently in three disposable cartridges) were further characterized concerning their molecular weight, hydrophobicity, and isoelectric point, shown in Figure 9. A two-way ANOVA could not determine statistical differences in the identified proteomes across the technical replicates. Finally, Figure 10 compares the number of identified peptides per protein across the three replicate sample preparations. The correlation coefficients in these graphs (0.94-0.95) demonstrate the high consistency of the sample preparation approach for bottom-up MS analysis.

Figure 1: Acetone-precipitated proteins. Samples containing 100 and 1,000 µg of protein combined with 100 mM NaCl and precipitated with 80% acetone (A) following 5 min precipitation time and (B) following precipitation and subsequent centrifugation. Please click here to view a larger version of this figure.

Figure 2: Protein precipitation by chloroform/methanol/water. A sample containing 50 µg of protein precipitated as per step 4. (A) Immediately following step 4.4. (B) Immediately following step 4.8. Please click here to view a larger version of this figure.
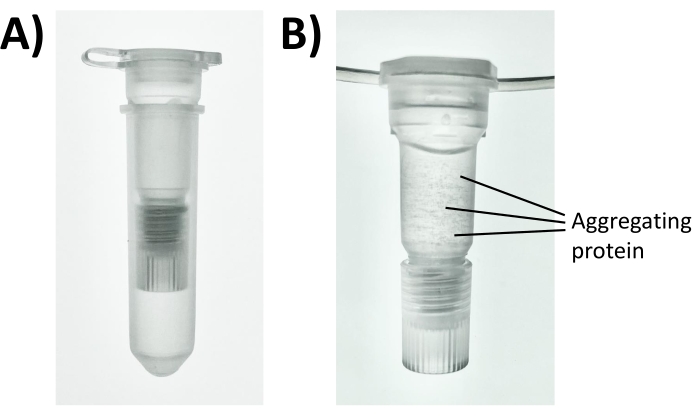
Figure 3: Photos of a disposable two-stage filtration and extraction cartridge for protein precipitation. A sample containing 100 µg protein was combined with 100 mM of NaCl and 80% acetone in (A) the assembled filtration and SPE cartridge and (B) precipitated for 5 min until protein aggregates became visible. Please click here to view a larger version of this figure.
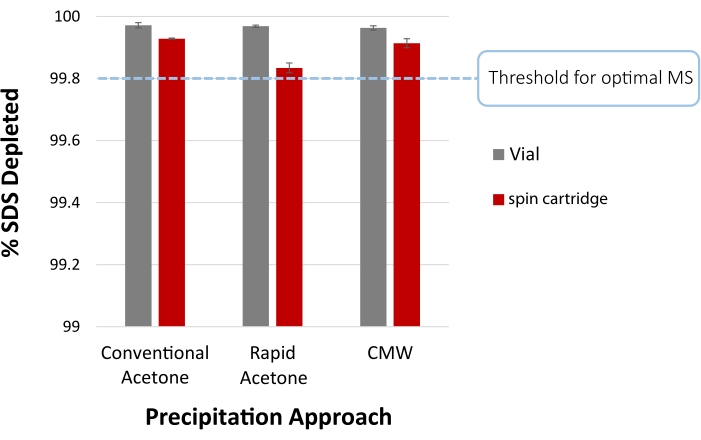
Figure 4: SDS depletion efficiency following protein precipitation. The percentage of SDS removed is shown from acetone precipitation with the conventional protocol (overnight at -20 °C), the rapid protocol (2 min incubation at room temperature), or by chloroform/methanol/water (CMW) precipitation of an S. cerevisiae lysate, both in conventional (vial) and cartridge format. These samples initially contained 0.5% SDS (5,000 ppm), inferring >99.8% SDS removal is required for optimal MS analysis. Residual SDS is quantified by methylene blue active substances (MBAS) assay. Error bars represent the standard deviation from technical replicates (n = 3). Please click here to view a larger version of this figure.
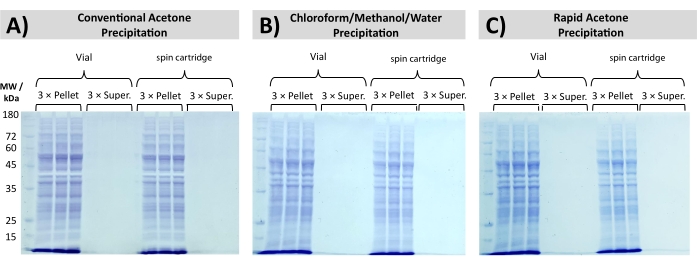
Figure 5: Total proteome recovery through precipitation. SDS PAGE shows the recovery of S. cerevisiae total protein lysate, precipitated by (A) conventional acetone precipitation, (B) chloroform/methanol/water precipitation, and (C) rapid acetone precipitation. Protein bands are exclusively observed in the pellet fraction, with no visible bands in the supernatant (Super.). Please click here to view a larger version of this figure.
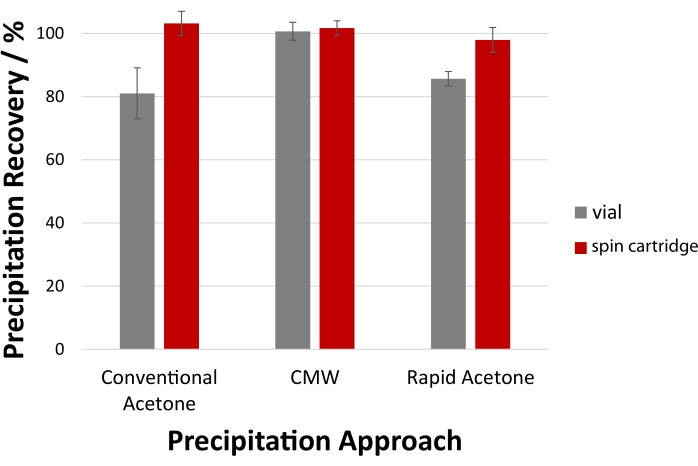
Figure 6: Superior protein recovery within a filtration cartridge. For precipitation of the S. cerevisiae total protein lysate, the disposable spin cartridge facilitates quantitative recovery with acetone and CMW precipitation. High recovery is also possible with vial-based precipitation, though careful sample manipulation and pipetting are required. LC-UV assessed protein recovery following resolubilization of the pellet with cold formic acid are shown here. Error bars represent the standard deviation from technical replicates (n = 3). Please click here to view a larger version of this figure.
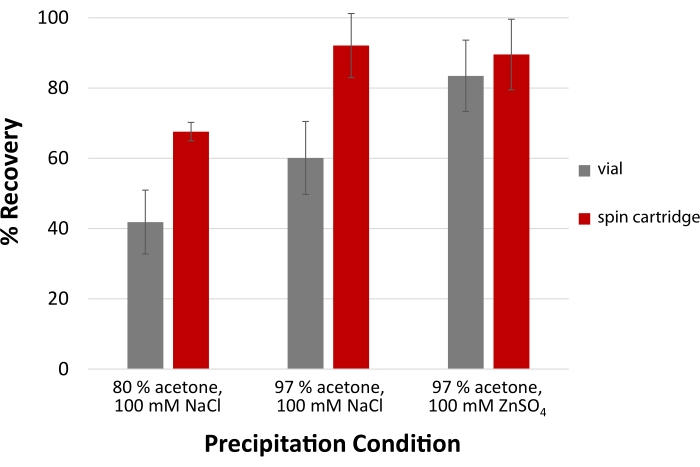
Figure 7: High precipitation yields for low molecular weight peptides. A modified acetone precipitation protocol for peptides and proteins ≤5 kDa involves coupling 100 mM of ZnSO4 with 97% acetone to achieve the highest yields. Precipitation facilitated by a disposable filtration cartridge demonstrates improved recovery compared to conventional vial-based precipitation across all three precipitation conditions. Error bars represent the standard deviation from technical replicates (n = 3). Please click here to view a larger version of this figure.
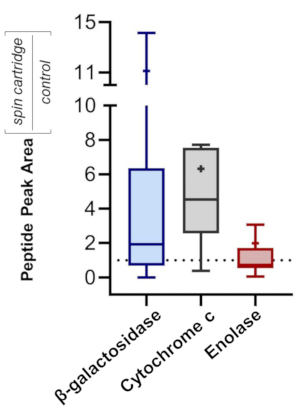
Figure 8: Higher recovery of standard proteins in SDS-based workflow. Tukey Box-and-Whisker plots30 of relative MS signal intensity for peptides recovered from SDS-containing proteins processed in a disposable filtration cartridge relative to a control sample (no SDS, no precipitation). The proteins employed span a wide concentration dynamic range-β-galactosidase:cytochrome c:enolase = 10,000:10:1. Each quartile within the boxes contains 25% of the distribution, while error bars encompass 95% of the distribution. Mean is indicated by "+" and median by a horizontal line. Please click here to view a larger version of this figure.
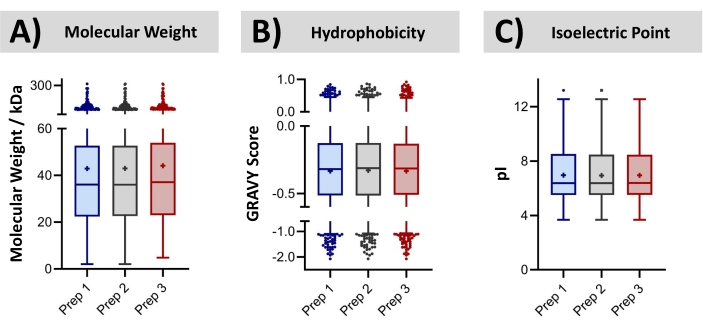
Figure 9: Identified protein distributions from technical replicates. Tukey Box-and-Whisker plots characterize (A) the molecular weight, (B) hydrophobicity, and (C) isoelectric point of proteins identified by bottom-up LC-MS/MS following triplicate preparations of a bovine liver lysate in a two-stage filtration and extraction cartridge. There was no statistical difference in these characteristics by two-way ANOVA (p < 0.05). Each quartile within the boxes contains 25% of the distribution, while error bars encompass 95% of the distribution. Please click here to view a larger version of this figure.
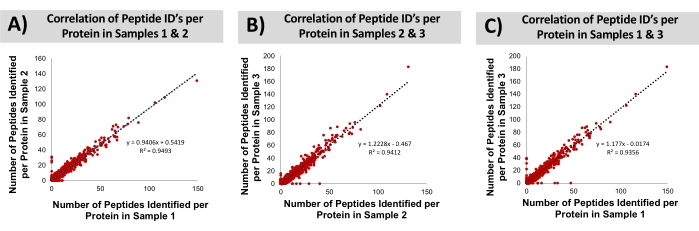
Figure 10: Correlation of peptide IDs per protein through the SDS-based preparation workflow across preparative replicates. Analysis of bottom-up proteome reproducibility across (A) samples 1 and 2, (B) samples 2 and 3, and (C) samples 1 and 3 based on the number of peptide MS identifications per protein. Please click here to view a larger version of this figure.
