Ethics approval and consent to participate: Human exfoliated deciduous dental pulp samples were received after obtaining informed consent and full ethical approval by Sri Rajiv Gandhi Dental College and Hospital (SRGCDS) Oral and Maxillofacial Department, Bengaluru, in accordance with the standards established by the Hospital Ethical Clearance Committee, SRGCDS. Following which isolation, culture, maintenance, and application of SHEDs were approved by and in compliance with the guidelines recommended by the Institutional Committee for Stem Cell Research (IC-SCR) at Manipal Institute of Regenerative Medicine, MAHE – Bengaluru. See the Table of Materials for details about all materials and reagents used in this protocol.
1. Preparation of reagents and buffers
- For culture maintenance
- Prepare MSC cell culture media (10%) using basal medium for undifferentiated hESCs, 10% fetal bovine serum (FBS), 1% L-glutamine, and 1% penicillin-streptomycin (Pen-strep) (Table 2).
- Prepare MSC cell culture media (20%) using basal medium for undifferentiated hESCs, 20% FBS, 1% L-glutamine, and 1% Pen-strep (Table 2).
- Prepare neutralizing media using basal medium for undifferentiated hESCs, 1% L-glutamine, and 1% antibiotic-antimycotic (Anti-Anti) (Table 2).
- For flow cytometric analysis and sorting
- Prepare staining buffer using 2% FBS in phosphate-buffered saline (PBS).
- Prepare a stock solution of 4′,6-diamidino-2-phenylindole (DAPI) (1 µg/mL) by adding 1 µL in 1 mL of PBS
- For lineage-specific differentiation of MSCs
- Prepare differentiation media for osteogenic, chondrogenic, and adipogenic differentiation according to the composition described in Table 3. Store the prepared aliquots at 4 °C for the duration of the experiment.
- Prepare serum-starved media (2% media) using basal medium for undifferentiated hESCs, 2% FBS, 1% L-glutamine, and 1% Pen-strep (Table 2).
| TYPE OF MEDIA | PURPOSE OF MEDIA | COMPOSITION FOR 50 mL | ||||||||
| FBS | Pen-Strep | L-Glutamine | BASAL MEDIUM FOR UNDIFFERENTIATED hESCs | |||||||
| 10% media | MSC culture and maintenance | 5 mL | 500 μL | 500 μL | 44 mL | |||||
| 20% media | CFU-F assay | 10 mL | 500 μL | 500 μL | 39 mL | |||||
| Serum starved (2%) media | Media for Control wells in Differentiation protocol | 1 mL | 500 μL | 500 μL | 48 mL | |||||
| Neutralizing media | Media for neutralizing the cell suspension after trypsinization | – | 500 μL | 500 μL | 49 mL | |||||
Table 2: Cell culture media for culture maintenance and assays. Abbreviations: MSC = mesenchymal stem cell; CFU-F = colony-forming unit-fibroblast.
| COMPONENTS | OSTEOGENIC MEDIA | CHONDROGENIC MEDIA | ADIPOGENIC MEDIA |
| Basal Media | 90 mL | 90 mL | 90 mL |
| Induction media | 10 mL | 10 mL | 10 mL |
| Total Volume | 100 mL | 100 mL | 100 mL |
Table 3: Differentiation media for trilineage differentiation of SHEDs.
2. Culture and maintenance of SHEDs
- Maintain cells in 10% MSC culture media and perform media changes every 2 days or as required.
- Trypsinize cells at 95% confluency using 0.25% trypsin-EDTA.
- Neutralize cells after trypsinization using neutralizing media.
- Centrifuge the tube at 300 × g for 6 min at room temperature to obtain a cell pellet.
- Decant the supernatant and resuspend the cell pellet in 10% MSC culture media.
- Seed cells into freshly prepared cell culture dishes containing 10% MSC culture media for further experiments or sub-culturing.
NOTE: Optimal seeding density for SHEDs is 0.2 × 106 cells in a 100 mm dish and 0.8 × 106 cells in a T-75 flask, to obtain 1.5 × 106 cells and 4 × 106 cells, respectively, at 90-100% confluency.
3. Characterization of MSCs
- Short-term cell growth assay
- Seed 4 × 104 cells/well in triplicate into 6-well plates in 10% MSC culture media.
- Incubate the plates for 7 days at 37 °C, and perform media changes every 2 days.
- Harvest the cells on days 2, 4, and 8 using 0.25% trypsin treatment and wash with culture media.
- Centrifuge the cells at 300 g for 6 min at room temperature and resuspend the pellet in 1 mL of media.
- Count the cells using a hemocytometer and determine their viability by the trypan blue exclusion method.
- Calculate the proliferation rate using equation (1):
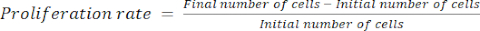 (1)
(1)
- Colony-forming unit-fibroblast (CFU-F) assay
- Seed 10,000 cells in a 100 mm dish and culture them in 20% MSC culture media.
- Incubate the plates for 14 days at 37 °C, and perform media changes every 3 days.
- After 14 days, rinse the colonies with PBS, fix them with crystal violet dye in methanol, and rinse again with PBS to remove the residual stain.
- Count and image the colonies.
NOTE: Count colonies with >50 cells. Colony-forming efficiency is calculated as colony-forming unit numbers.
- Immunofluorescence assay
- Seed the MSCs on 35 mm dishes and let them grow till 80-90% confluency.
- Remove the media and rinse the dishes with PBS once.
- Fix the cells with 1 mL of 4% paraformaldehyde (PFA) by either incubating for 1 h at room temperature or overnight at 4 °C.
- After fixation, wash the wells with PBST for 3 x 5 min on the rocker.
- Add 0.3% Triton X-100 in PBST (0.05% Tween 20 solution in PBS) for permeabilizing the cells. Keep it on the rocker for 15 min at room temperature.
- Wash the wells with PBST for 3 x 5 min on the rocker.
- Add 3% bovine serum albumin (BSA) for blocking and keep it on the rocker for 1 h at room temperature.
- Wash the wells with PBST for 3 x 5 min on the rocker.
- Add 800 µL of 1:500 dilution of mouse anti-vimentin, keep it on the rocker for 1 h at room temperature, and transfer the plate to 4 °C for overnight incubation.
- Next day, remove the primary antibody and wash the wells with PBST for 3 x 5 min on the rocker.
- Add 800 µL of 1:1,000 dilution of goat anti-mouse IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 555, and keep it for 3 h at room temperature on the rocker.
- Wash the wells with PBST for 3 x 5 min on the rocker.
- Add Alexa fluor 488 Phalloidin Probes 240 µL in 1,000 µL of PBS and incubate at room temperature for 60 min on the rocker.
- Wash the wells with PBST for 3 x 5 min on the rocker.
- Add 700 µL of DAPI mountant and observe the cells under a microscope.
- Lineage-specific differentiation
- Differentiation following 2D culture
- Take two 48-well plates and label them as osteogenic and adipogenic lineages, respectively.
- Seed 15,000 cells/well in four wells of each plate and culture them in 10% MSC culture media.
- Once the monolayer of cells has reached 90% confluency, label the first two wells as 'Control' and replace the existing media with serum-starved media (2% media). In the last two wells labeled as 'Test,' add differentiation media of either of the two lineages, adipogenic or osteogenic. Mark this as Day 0.
- Gently replace media every third day to circumvent peeling and be careful to avoid contamination.
- Maintain these conditions till the twenty-first day; then process for the staining experiment.
- Differentiation following 3D culture
- Take two 15 mL tubes for performing chondrogenic differentiation using 3D pellet culture.
- Transfer 1 × 106 cells to each tube and centrifuge at 300 × g for 6 min to form a pellet. Label one tube as 'Control' and add 10% MSC culture media to it; label the other tube as 'Test' and add chondrogenic differentiation media. Place the tubes carefully in the incubator with their caps loosely screwed. Mark this as Day 0.
- Change the media every third day carefully, so as not to dislodge/disintegrate the pellet during media changes.
- Maintain these conditions till the twenty-first day and then, process the cells for further experiments.
- Differentiation following 2D culture
- Lineage-specific cytochemical staining
- For 2D cultures, use the differentiated (test) and undifferentiated MSCs (control) plates (from step 3.4.1.5.) for staining, and first, remove the media and wash twice with PBS. Fix the cells using 4% PFA for 30 min at room temperature, remove the supernatant, and wash once with PBS. Perform staining for each lineage as follows.
- For adipocyte lineage, add permeabilization solution from the kit and incubate the plate for 5 min at room temperature. Prepare and add 1 mL of Oil red O working solution and keep it for 10 min. Remove the stain and give five washes with distilled water.
- For osteocyte lineage, add 5% of freshly prepared silver nitrate (in distilled water) to each well and keep the plate under UV for 1 h. Remove the solution and add 2.5% of sodium thiosulphate to remove the unreacted silver; keep it for 5 min. Remove the stain, wash twice with distilled water, and observe the stained cells under a microscope.
- For 3D cultures, collect the pellet after the end of the differentiation period from step 3.4.2.3 and obtain cryosections of the differentiated tissue in the form of the pellet. Allow the slides to air dry and be at room temperature before proceeding to the staining.
- For chondrocyte lineage, follow the directions of the staining kit to add a sufficient volume of washing solution, remove it, add the fixing solution, and incubate for 30 min. Wash with distilled water, add the staining solution, and incubate for 30 min. Wash thrice with 0.1 N hydrochloric acid; add distilled water to neutralize the acidity. Observe the stained cells under a brightfield microscope.
- For 2D cultures, use the differentiated (test) and undifferentiated MSCs (control) plates (from step 3.4.1.5.) for staining, and first, remove the media and wash twice with PBS. Fix the cells using 4% PFA for 30 min at room temperature, remove the supernatant, and wash once with PBS. Perform staining for each lineage as follows.
4. Cell surface staining for immunophenotyping
NOTE: Recommended cell culture plates for getting an optimal number of cells in steps 4.2-4.5 are 100 mm dishes or T75 flasks.
- Cell preparation for flow cytometric experiments
- Trypsinize and collect the cells from a confluent dish/ flask and centrifuge at 300 × g for 6 min to obtain the cell pellet.
- Resuspend the pellet in 1 mL of media and determine the viable cell count using a hemocytometer following the trypan blue exclusion method.
- Centrifuge the cell suspension again after counting and give two more washes to the pellet with 1 mL of staining buffer.
- Discard the supernatant and finally resuspend the pellet in an appropriate volume of the staining buffer depending on the protocol (see steps 4.2 to 4.5).
- Preparation of compensation controls
- Take seven FACS tubes and label them as Unstained, DAPI, V450, FITC, PE, PerCP-Cy 5.5, and APC.
- Resuspend the final pellet in the staining buffer (see step 4.1.) keeping a cell density of 0.5 × 106 cells per 50 µL per tube for the Unstained and DAPI tubes.
- Prepare the single-stained tubes for compensation as described in Table 4.
- Gently vortex each tube after preparation and incubate in the dark for 30 min.
- After incubation, give two washes by adding 1 mL of staining buffer to each tube, followed by brief vortexing and centrifugation at 200 g for 10 min at room temperature.
- Discard the supernatant, resuspend the pellet in 500 µL of staining buffer, and set it aside until the acquisition.
- For the DAPI tube, perform heat shock treatment by incubating it in a 60 °C water bath for 5 min followed by 15 min on ice. Add 5 µL of DAPI to the suspension and keep it in the dark until the run acquisition.
- Preparation of fluorescence minus one (FMO) controls
- Resuspend the final pellet in staining buffer (see step 4.1) keeping a cell density of 0.5 × 106 cells per 50 µL for each tube.
- Take five FACS tubes, and label them as CD44-V450 isotype, CD90-FITC, CD45-PE, CD73-PerCP Cy5.5 isotype, and CD105-APC isotype.
- Prepare the cell and antibody suspension according to Table 5.
- Vortex the tubes gently and incubate them for 30 min at room temperature in the dark.
- After incubation, give two washes with 1 mL of staining buffer to each tube followed by brief vortexing and centrifugation at 200 g for 10 min at room temperature.
- Discard the supernatant, resuspend the pellet in 500 µL of staining buffer, and set it aside until acquisition.
- Preparation of samples for analysis
- Resuspend the final pellet in the staining buffer (see step 4.1) keeping a cell density of 0.5 × 106 cells per 50 µL for each tube.
- Take two FACS tubes and label them as Mixed tubes 1 and 2.
- Prepare the cell and antibody suspension according to Table 6.
- Vortex the tubes gently and incubate them for 30 min at room temperature in the dark.
- After incubation, give two washes with 1 mL of staining buffer to each tube followed by brief vortexing and centrifugation at 200 g for 10 min at room temperature.
- Discard the supernatant, resuspend the pellet in 500 µL of staining buffer, and set aside until acquisition.
- Preparation of samples for single-cell sorting
- Take two FACS tubes, add 1mL of FBS, and roll the tube around till an even layer of FBS is formed on the inside of each tube. Incubate this for 1-2 h while processing the sample for staining. Label these tubes as Mixed tubes 1 and 2.
- Resuspend the final pellet in the staining buffer (see step 4.1) keeping a cell density of 2-3 × 106 cells per 50 µL for each tube.
- Prepare the cell and antibody suspension in Mixed tubes 1 and 2 according to Table 7.
- Vortex the tubes gently and incubate them for 30 min at room temperature in the dark.
- After incubation, give two washes with 1 mL of staining buffer to each tube followed by brief vortexing and centrifugation at 200 g for 10 min at room temperature.
- Discard the supernatant, resuspend the pellet in 500 µL of staining buffer, and set aside. Add 5 µL of DAPI 15 min prior to sorting.
NOTE: Note that for sorting experiments higher cell density is recommended per tube.
| Tube type | Positive Comp Beads* | Negative Comp Beads* | Cells | Antibody added | |
| FITC tube | 1 drop | 1 drop | – | Anti-human CD90-FITC (2 µL) | |
| V450 tube | 1 drop | 1 drop | – | Anti-human CD44-V450 (2 µL) | |
| PerCP-Cy 5.5 tube | 1 drop | 1 drop | – | Anti-human CD73-PerCP Cy 5.5 (2 µL) | |
| PE tube | 1 drop | 1 drop | – | Anti-human CD45-PE (2 µL) | |
| APC tube | 1 drop | 1 drop | – | Anti-human CD105-APC (2 µL) | |
| DAPI tube | – | – | 50 µL | – | |
| Unstained tube | – | – | 50 µL | – | |
| *1 drop= 60 µL of bead suspension | |||||
Table 4: Compensation control samples. Abbreviations: Comp = compensation; DAPI = 4',6-diamidino-2-phenylindole; FITC = fluorescein isothiocyanate; APC = allophycocyanin; PE = phycoerythrin; PerCP = peridinin-chlorophyll-protein.
| TUBE TYPE | ANTIBODY AGAINST POSITIVE MARKER (2 µL) | ANTIBODY AGAINST NEGATIVE MARKER (2 µL) | ISOTYPE ANTIBODIES ADDED (2 µL) | TOTAL VOLUME OF ANTIBODIES | VOLUME OF CELL SUSPENSION ADDED | VOLUME OF STAINING BUFFER ADDED | ||
| CD90-FITC FMO tube | – Anti-human CD44-V450 | Anti-human CD45-PE | FITC IgG1 isotype | 10 µL | 50 µL | 40 µL | ||
| – Anti-human CD73 PerCP Cy 5.5 | ||||||||
| – Anti-human CD105-APC | ||||||||
| CD73-PerCP Cy5.5 FMO tube | – Anti-human CD44-V450 | Anti-human CD45-PE | PerCP Cy 5.5 IgG1 isotype | 10 µL | 50 µL | 40 µL | ||
| – Anti-human CD90-FITC | ||||||||
| – Anti-human CD105-APC | ||||||||
| CD44-V450 FMO tube | – Anti-human CD90-FITC | Anti-human CD45-PE | V450 IgG1 isotype | 10 µL | 50 µL | 40 µL | ||
| – Anti-human CD73-PerCP Cy 5.5 | ||||||||
| – Anti-human CD105-APC | ||||||||
| CD105-APC FMO tube | – Anti-human CD44-V450 | Anti-human CD45-PE | APC IgG1 isotype | 10 µL | 50 µL | 40 µL | ||
| – Anti-human CD90-FITC | ||||||||
| – Anti-human CD73-PerCP Cy 5.5 | ||||||||
| CD45-PE FMO tube | – Anti-human CD44-V450 | – | PE IgG1 isotype | 10 µL | 50 µL | 40 µL | ||
| – Anti-human CD90-FITC | ||||||||
| – Anti-human CD73-PerCP Cy 5.5 | ||||||||
| – Anti-human CD105-APC | ||||||||
Table 5: FMO control samples. Abbreviations: FMO = fluorescence minus one; FITC = fluorescein isothiocyanate; APC = allophycocyanin; PE = phycoerythrin; PerCP = peridinin-chlorophyll-protein.
| TUBE TYPE | ANTIBODY AGAINST POSITIVE MARKER (2 µL) | ANTIBODY AGAINST NEGATIVE MARKER (2 µL) | TOTAL VOLUME OF ANTIBODIES | VOLUME OF CELL SUSPENSION ADDED | VOLUME OF STAINING BUFFER ADDED | |
| Mixed tube 1 | – Anti-human CD44-V450 | Anti-human CD45-PE | 10 µL | 50 µL | 40 µL | |
| – Anti-human CD90-FITC | ||||||
| – Anti-human CD73-PerCP Cy5.5 | ||||||
| – Anti-human CD105-APC | ||||||
| Mixed tube 2 | – Anti-human CD44-V450 | Anti-human CD45-PE | 10 µL | 50 µL | 40 µL | |
| – Anti-human CD90-FITC | ||||||
| – Anti-human CD73-PerCP Cy5.5 | ||||||
| – Anti-human CD105-APC | ||||||
Table 6: Sample tubes for multicolor immunophenotyping of SHEDs. Abbreviations: SHEDs = stem cells from human exfoliated deciduous teeth; PE = phycoerythrin.
| TUBE TYPE | ANTIBODY AGAINST POSITIVE MARKER (3 µL) | ANTIBODY AGAINST NEGATIVE MARKER (3 µL) | TOTAL VOLUME OF ANTIBODIES | VOLUME OF CELL SUSPENSION ADDED | VOLUME OF STAIN BUFFER ADDED | |
| Mixed tube 1 | – Anti-human CD90-FITC | Anti-human CD45-PE | 9 µL | 50 µL | 41 µL | |
| – Anti-human CD73-PerCP Cy5.5 | ||||||
| Mixed tube 2 | – Anti-human CD90-FITC | Anti-human CD45-PE | 9 µL | 50 µL | 41 µL | |
| – Anti-human CD73-PerCP Cy5.5 | ||||||
Table 7: Single-cell sorting reaction tubes. Abbreviations: FITC = fluorescein isothiocyanate; PE = phycoerythrin; PerCP = peridinin-chlorophyll-protein.
5. Single-cell sorting
- Preparation of cell sorter
- Install a 100 µm nozzle in the sorter.
NOTE: The appropriate nozzle is at least five times the diameter of the particle to be sorted. The sheath fluid to be used for sorting needs to be decided based on the sample type and the sensitivity of the experiment; for this experiment, proprietary sheath fluid has been used. - Perform the daily instrument quality check (QC) and set up the sorter for the experiment. Refer to the instrument manual for a detailed guide to instrument setup.
- Install a 100 µm nozzle in the sorter.
- Setting up the compensation matrix
- Set the compensation matrix using single-stain compensation tubes from step 4.2.
- In the proprietary software, select Experiment from the Toolbar and click on Compensation setup. Open Create compensation controls.
- Check the markers and confirm. A new specimen is added named "Compensation" under which new tubes named the marker controls are added automatically by the software.
- Select the Unstained tube and run it, to record 5,000 events. Drag the gate to the population of cells and apply it to all compensation controls. This is to set the voltages and negative gate for each fluorescent parameter.
- Similarly, load the single-stain compensation tubes separately, and record and save the data. Select the gate demarcating the population of interest and apply to all compensation controls. This is to set the positive gates for each fluorescent parameter.
- Select Experiment from the Toolbar and click on Calculate compensation values | link and save.
NOTE: Once the auto-comp matrix is generated using the software, the voltage parameters of the fluorochromes cannot be changed for any of the channels in the Mixed tubes.
- Data acquisition
- Record 10,000 cells in each tube from steps 4.3. and 4.4 to collect data for analysis of the immunophenotype of the cells.
- Preparation of the collection devices
- Depending on the purpose of the sorted cell populations, choose between 6-well, 24-well, 48-well, or 96-well plates.
- Coat the wells with 200-500 µL of FBS and keep the plates undisturbed for 2 h.
- After 2 h, remove the residual FBS and add 200-500 µL of 10% MSC culture media.
- Cell sorting in single-cell sort mode
- Run Mixed tube 1 (from step 4.5), and record 10,000 events to set the gates on the population of interest to be sorted, using the appropriate gating strategy.
- Load a collection plate and set target cell numbers between 2,500 and 5,000 cells/well and select the single-cell sort purity mask.
- Collect the sorted populations in the collection device and keep them on ice until the end of the sorting experiment.
- Once done, transfer the plates to the 5% CO2 incubator to maintain the cultures at 37 °C.
NOTE: Post acquisition, raw data files were exported as .fcs file format (v.3.0. onwards). The sort reports generated after every experiment recorded the number of events/cells sorted per assigned well and indicated the number of conflicts that were aborted.
The SHEDs were characterized with standard immunofluorescence assays showing the expression of vimentin (red, type III intermediate filaments), actin filaments (Alexa fluor 488 Phalloidin Probes), and nuclei stained with DAPI (Figure 1A). To estimate their proliferative and colony-forming capacities, standard short-term cell growth assays were performed. A 14.3-fold increase in proliferation rate from day 2 to day 8 has been shown in Figure 1B. The clonogenic properties of the SHEDs were determined from the CFU-F Assays shown in Figure 1C–E. As per the ISCT criteria, the multipotent MSCs were able to differentiate into the three mesodermal-derived lineages. Cytochemical staining with Oil Red O was used to detect the oil droplets in the differentiated adipocytes (Figure 1F); von Kossa staining was used to stain the calcium deposits found in the matrix of the differentiated osteoblasts (Figure 1G); and the aggrecans in the 3D culture were stained for Alcian Blue in the differentiated chondroblasts (Figure 1H).
To characterize the immunophenotype of the SHEDs, multiparametric flow cytometry assays were set. The experiments were performed along with the use of four types of experimental controls, namely compensation controls (Table 4), FMO and isotype controls (Table 5), and unstained controls. The isotypes of all the MSC and HSC antibodies used were added along with the FMO tubes to discriminate positive versus negative signals and high versus dim antigen expression. Figure 2A shows the distribution of the FMO and isotype controls used per experiment. The representative density plots showed no expression of the FITC isotype of CD90 FITC antibodies. Similarly, the histogram overlays (Figure 2B) compared the expression levels of the stained samples along with their controls in the respective channels of FITC, PerCP Cy5.5, and PE. Positive expression of the markers CD90 and CD73 (red histograms) and negative expression of CD45 are comparable with that of their unstained and FMO controls (blue and black histograms, respectively).
Flow cytometry-based characterization of the immunophenotypes has shown the marker distribution from one of the early passages (P6) SHEDs (Figure 3A–H). Both the ISCT-recommended markers and CD44 determined the parent populations as MSCs. The tetra-positive population demonstrated in the density plots showed ≥98% expression of CD90+CD73+CD105+CD44+ and ≤2% expression of CD45 markers. More than five independent experiments showed similar results. The late-passage SHEDs (P12), which showed differential expression of the positive markers, were chosen for single-cell sorting of high and dim expressors (Figure 4A–H). Following the gating strategy (Figure 4H), singlets→ Live cells→ Non-HSCs→ Double positive CD73+CD90+ and single positive CD73+CD90– populations were sorted using the single-cell sort mode. The sorted populations were collected in 96-well/48-well/6-well plate layouts with different seeding counts per well (Supplementary File 1). Three consecutive attempts were made in a single phase of sorting experiments and the survivability of cells post sort (in the double-positive expressors: CD90+CD73+) per experiment was 100%. Three independent phases of experiments have been conducted. We matched the number of cells that grew per well post sort to the number of cells sorted per well.
Figure 5A shows the CD90+CD73+ post-sorted cells collected in the 96-well plate showed adherence and proliferation like their parent population. The sorted cells have been observed to reach 70-80% confluency on the sixth day post sorting. The adhered and proliferating post-sorted cells were differentiated in the presence of adipogenic growth factors and then stained post Day 15 with Oil Red O using appropriate post-sorted control (undifferentiated) cells as well (Figure 5B,C).
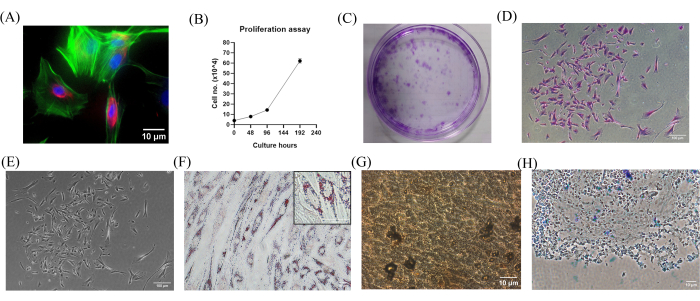
Figure 1: Characterization of stem cells from human exfoliated deciduous teeth. (A) Immunofluorescence staining confirms that the isolated MSCs express vimentin (red, type III intermediate filaments), actin filaments (Alexa fluor 488 Phalloidin Probes), and DAPI (blue, nuclei). Photomicrograph obtained at original magnification of 20x. Scale bar = 10 µm. (B) Short-term cell growth assay of SHEDs exhibits their high proliferative capacity, as observed by the significant increase in the number of cells in culture by day 2 and a 14.3-fold increase in proliferation rate at the end of day 8 (n = 3, p-value<0.01). Standard deviation error bars have been shown. (C–E) CFU-F assay and Crystal violet staining confirm the colony-forming ability of SHEDs; (C) multiple colonies are expected to form after 14 days in culture; (D) single clonogenic colony originating from a single cell; (E) Crystal violet-stained single colony of SHEDs displays the typical fibroblast-like morphology of the cells. (F–H) MSCs under appropriate in vitro conditions are expected to undergo differentiation into adipogenic, osteogenic, and chondrogenic lineages by day 21. Cytochemical staining shows (F) Oil Red O (inset shows 40x image of adipocyte showing oil droplets), (G) von Kossa, and (H) Alcian Blue staining for adipogenic, osteogenic, and chondrogenic lineages, respectively, at Day 22. Photomicrographs are obtained at an original magnification of 20x. Scale bar = 10 µm. Abbreviations: MSCs = mesenchymal stem cells; DAPI = 4',6-diamidino-2-phenylindole; SHEDs = stem cells from human exfoliated deciduous teeth; CFU-F = colony-forming unit-fibroblast. Please click here to view a larger version of this figure.
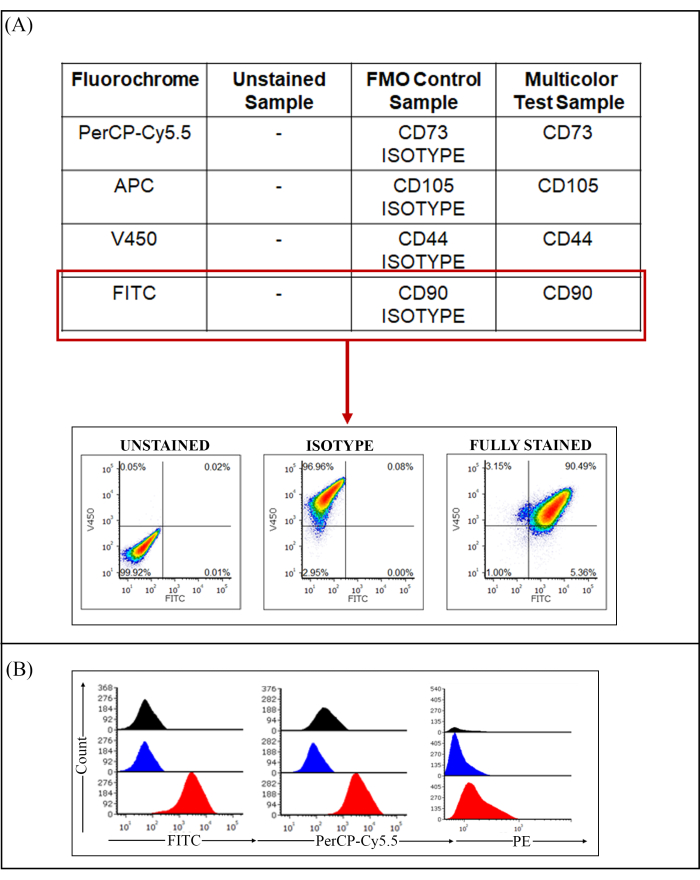
Figure 2: Negative controls used in multiparametric experiment. (A) Fluorescence Minus One controls have been displayed in a tabular form, where FMO controls have been replaced by their specific isotypes. The representative density plots (left to right) show the unstained control (left), FMO of CD90 FITC (middle) with all antibodies added except CD90 FITC along with the addition of CD90 FITC isotype, and fully stained sample with all antibodies of the panel added (right). (B) Histogram overlay represents the comparison of three types of samples, black represents FMO, blue represents unstained, and red represents fully stained samples in their respective channels. Abbreviations: FMO = Fluorescence Minus One; FITC = fluorescein isothiocyanate; PerCP = peridinin-chlorophyll-protein; PE = phycoerythrin. Please click here to view a larger version of this figure.
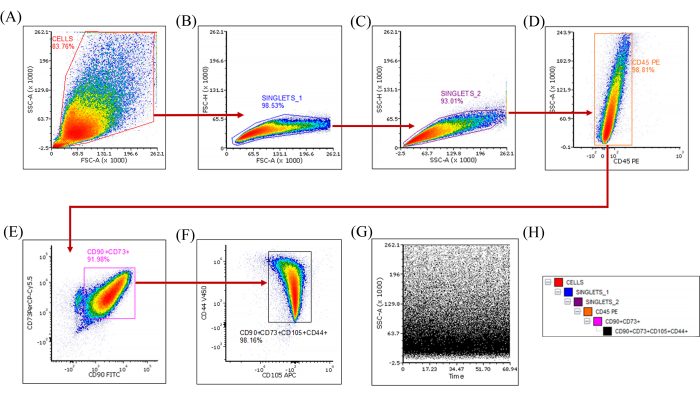
Figure 3: Multiparametric immunophenotyping of SHEDs (P6). (A) SSC-A versus FSC-A: Based on their side and forward scatter properties, cells of interest are gated and separated from the debris; (B) FSC-H versus FSC-A and (C) SSC-H versus SSC-A:The two plots are employed for doublet discrimination, which enables the selection of singlets while eliminating aggregates that may generate false positive signals; (D) SSC-A versus CD45 PE: Exclusion gating allows selection of the CD45– population from the singlets; (E) CD73 PerCP-Cy5.5 versus CD90 FITC: From the CD45– live singlets, CD90+CD73+ cells are gated; (F) CD44 V450 versus CD105 APC: The CD90+CD73+ population, is further gated to define the tetra-positive CD90+CD73+CD44+CD105+ cells; (G) SSC-A versus Time(s): The plot depicts the stable acquisition over time (seconds) and that no events caused pressure fluctuations while acquisition; (H) Gating hierarchy. Abbreviations: SHEDs = stem cells from human exfoliated deciduous teeth; FITC = fluorescein isothiocyanate; APC = allophycocyanin; PE = phycoerythrin; PerCP = peridinin-chlorophyll-protein; SSC-A = side scatter-peak area; FSC-A = forward scatter-peak area; FSC-H = forward scatter-peak height. Please click here to view a larger version of this figure.
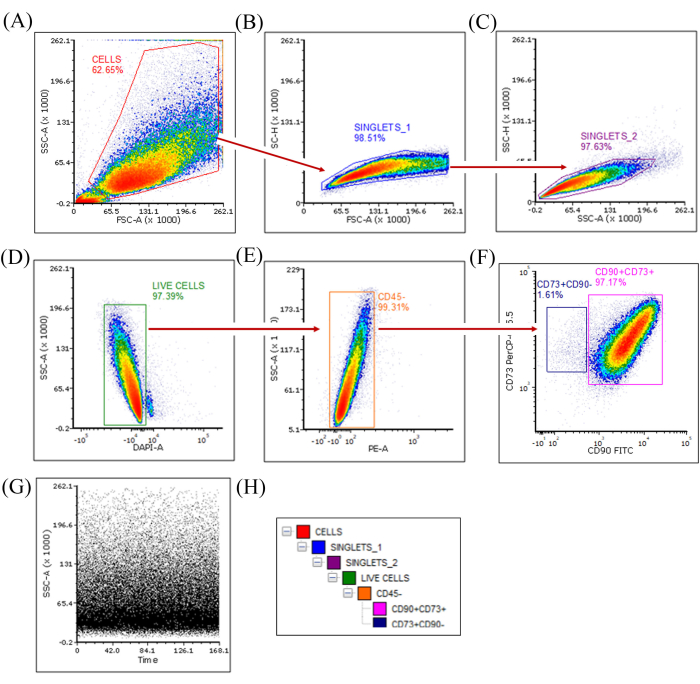
Figure 4: Single-cell sorting of immunophenotyped SHEDs (P12). (A–F) A step-by-step exclusion-based gating strategy to sort pure MSCs in the following sequential order: Cells to singlets_1, to singlets_2, to DAPI– live cells, to CD45– non-Hematopoietic cells, and finally to CD90+CD73+ cells. The gated populations shown in (F) were sorted using single-cell sort mode. (G) The SSC-A versus Time(s) plot describes the uninterrupted sorting of the samples. (H) Gating hierarchy. (n=3, Sort efficiency = 100%). Abbreviations: SHEDs = stem cells from human exfoliated deciduous teeth; MSCs = mesenchymal stem cells; DAPI = 4',6-diamidino-2-phenylindole; FITC = fluorescein isothiocyanate; APC = allophycocyanin; PE = phycoerythrin; PerCP = peridinin-chlorophyll-protein; SSC-A = side scatter-peak area. Please click here to view a larger version of this figure.
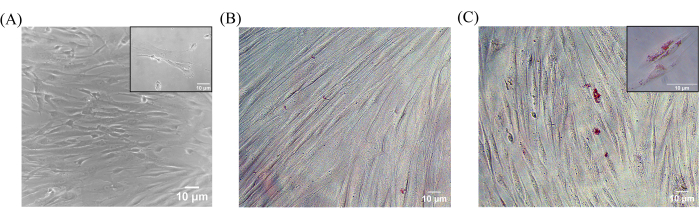
Figure 5: Morphological and functional characterization of post-sorted SHEDs. (A) Phase contrast images of post-sorted populations were collected in a 96-well plate on Day 6 at an original magnification of 10x, Scale bar = 10 µm (inset shows a 20x image depicting the sorted single cells changing morphology into fibroblast-like MSCs). Cytochemical staining for adipogenic differentiation using Oil Red O observed in (B) undifferentiated (control) and (C) differentiated post-sort population at an original magnification of 20x, Scale bar = 10 µm (inset shows a 40x image depicting the oil droplets observed). Abbreviations: SHEDs = stem cells from human exfoliated deciduous teeth. Please click here to view a larger version of this figure.
Supplementary File 1: Compiled sort reports generated per experiment. Please click here to download this File.
Supplemental Table S1: Optical Configuration of BD FACSAria Fusion. Abbreviations: LP = long pass; PMT = photomultiplier tube; DAPI = 4',6-diamidino-2-phenylindole; FITC = fluorescein isothiocyanate; APC = allophycocyanin; PE = phycoerythrin; PerCP = peridinin-chlorophyll-protein. Please click here to download this File.
