In order to determine if an intercalating dye will affect a restriction enzyme digesting DNA, the correct order of steps must be followed (Figure 1). Once DNA is stained and digested with a given enzyme, a picture of the gel can be taken to determine the number of fragments and their size (Figure 2). In order to determine the enzyme efficiency, the total number of expected visible bands divided by the number of visible bands. Enzyme efficiency equaled unity, if the number of bands expected and seen match. If there are more bands on the gel then expected, the value is greater than one and indicates partially digested reactions. If there are less bands then expected, then the value is less than one, which indicates incomplete digestion. In Figure 2a, lanes 3 and 4 show the mobility of the bands has decreased, causing the bands to shift towards the wells. In lanes 1 and 2, the amount of dye does not affect the mobility noticeably, so the bands match the control. In Figure 2a, lanes 5 and 6 show more bands than the control, so it indicates partial digestion. Which means, the dyes affected the cleavage of the DNA at the recognition site for that enzyme. In Figure 2b, all expected bands are seen for all dye concentrations, so the dye did not interfere with the digestion of DNA. Figure 2c tracks the mobility shifts with colored asterisks.
As dye concentration increases for dyes with -1, the mobility decreases. In order to determine the approximate size of the bands, a control is required to know where the native DNA molecules are expected compared to the stained DNA (Figure 2a). For dyes with -3, the mobility did not decrease to the extent that -1 dyes did, so it was easier to determine the fragment size (Figure 2b). The mobility difference is due to the type of dye utilized, as seen in Maschmann et al.25.The variance of these dyes is seen due to the structures of the dyes. The linker between the aromatic moiety for dyes -3 was longer than -1. The dyes may intercalate slightly differently due to the difference in the linker size, which may explain why the mobility shift is less for -3 dyes.
If there is no DNA, or the DNA bands are very faint in the gel, increasing the concentration of DNA is required. However, if the DNA concentration increases, then the amount of dye required will also increase to keep the correct concentration of dye to DNA ratio.
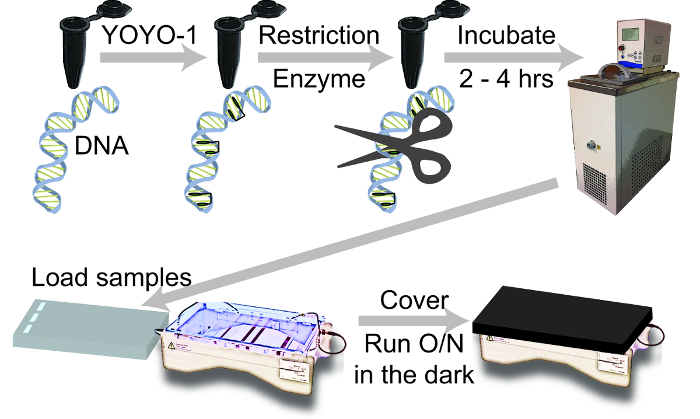
Figure 1: A schematic of the steps required when digesting fluorochrome labeled DNA molecules. DNA and YOYO-1, or another dye from the TOTO series, is added to a black tube and incubated overnight (O/N; 15 – 20 h). A restriction enzyme is added to a tube; it is placed in an incubator, at the preferred temperature, for 2 – 4 h (Table 2). Load samples into an immersed 0.7% agarose gel made with 1x TAE buffer. Dark colored paper covers the gel box and the lights are turned off, while the gel is ran overnight to prevent photocleavage of the dye. Please click here to view a larger version of this figure.
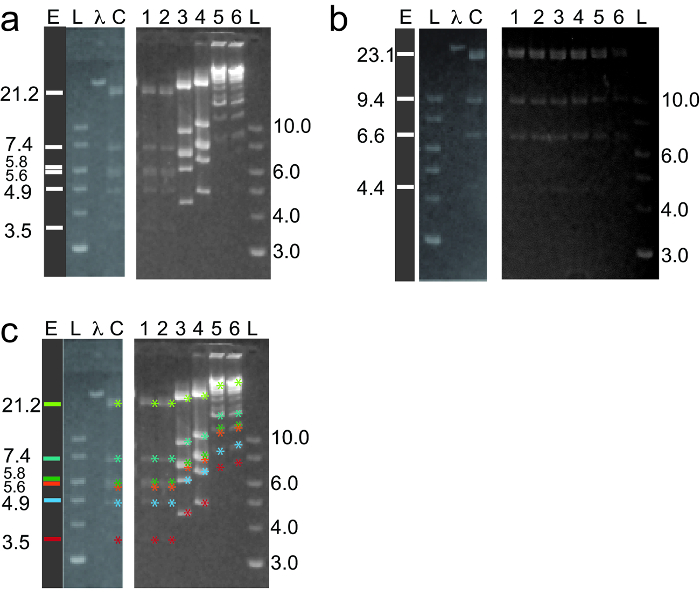
Figure 2: Restriction digestion of DNA intercalated with a cyanine dimer dye. (a) Lambda DNA is stained with different concentrations of YOYO-1 overnight (lane C: digested lambda DNA with no dye (control), lane 1: 1.9 ng, lane 2: 3.8 ng, lane 3: 19.4 ng, lane 4: 32.2 ng, lane 5: 48.3 ng, lane 6: 63.5 ng of dye per 100 ng of DNA) and then digested with EcoRI at 37 °C for 2 – 4 h. All reactions are electrophoresed (E = 0.85 V/cm) on a 0.7% high gelling temperature (HGT) agarose gel made with 1x TAE buffer (1x TAE; 40 mM Tris-HCl, 20 mM Acetic acid, 1 mM EDTA). Gels are stained with ethidium bromide and imaged with a blue light transilluminator coupled to a camera. Lane L is a 1 kb ladder (sizes, kb); lane λ is full length lambda DNA; lane E is the expected band pattern (sizes, kb). (b) Lambda DNA is stained with YOYO-3 (lane 1: 1.9 ng, lane 2: 3.8 ng, lane 3: 19.4 ng, lane 4: 32.2 ng, lane 5: 48.3 ng, lane 6: 63.5 ng of dye per 100 ng of DNA) and digested with HindIII at 37 °C. (c) To see how the mobility shifted as the YOYO-1 concentration is increased, colored asterisks are located next to each band for each YOYO-1 concentration. For example, the band at 3.5 kb has a red asterisk next to each band in each YOYO-1 concentration. The unexpected bands in lane 5 and 6 do not have an asterisk next to the bands. The figure is modified from Maschmann et al.25. Copyright (2017) Nucleosides, Nucleotides, and Nucleic Acids. Please click here to view a larger version of this figure.
| Chemical Name | Abbreviated Name | Ex/Em (nm) |
| [(1–1′-[1,3-propanediylbis[(dimethyliminio)-3,1-propanediyl] bis[4-[(3-methyl-2(3H)-benzothiazolylidene)methyl]]-, tetraiodide | TOTO-1 | 514/533 |
| 1,1′ – (4,4,8,8-tetramethyl-4,8-diazaundecamethylene)bis[4-[(3-methylbenzo-1,3-oxazol -2-yl)methylidene]-l,4-dihydroquinolinium] tetraiodide} | YOYO-1 | 491/509 |
| 2,2′-[1,3-prop-anediylbis[(dimethyliminio)-3,1-propanediyl-1(4H)-pyridinyl-4-ylidenemethylid- yne]]bis[3-methyl]-, tetraiodide | POPO-1 | 434/456 |
| 2,2′-[1,3-propanediylbis[(dimethylimi- nio)-3,1-propanediyl-1(4H)-pyridinyl-4–1,1′ -[4,8-bis(dimethyliminio)undecane-1,11-diyl]bis{4-[3-(3-methyl-1,3-benzothiazol-2(3H)-ylidene)prop-1-en-1-yl]qui- nolinium} | BOBO-1 | 462/481 |
| 2,2′-{[4,8-bis(dimethyliminio)undecane-1,11-diyl]bis[quinolin-1-yl-4- ylideneprop-1-en-1-yl-3-ylidene]}bis(3-methyl-1,3-benzothiazol-3-ium) tetraio- dide | TOTO-3 | 642/660 |
| 1,1′-[1,3-propanediylbis[(dimethyliminio)-3,1-propanediyl]]bis[4- [3-(3-methyl-2(3H)-benzoxazolylidene)-1-propenyl]]-, tetraiodide | YOYO-3 | 612/631 |
| 2,2′ -[1,3-propanediylbis[(dimethyliminio)-3,1-propanediyl-1(4H)-pyridinyl-4- ylidene-1-pr-open-1-yl-3-ylidene]]bis[3-methyl]-, tetraiodide | POPO-3 | 534/570 |
| 2,2'-{propane-1,3-diylbis[(dimethylazaniumdiyl)propane-3,1-diylpyridin-1-yl-4-ylideneprop-1-en-1-yl-3-ylidene]}bis(3-methyl-1,3-benzothiazol-3-ium) tetraiodide | BOBO-3 | 570/602 |
Table 1: Abbreviated names of TOTO family of dyes with IUPAC names with emission (Em) and excitation (Ex) maximima22.
| Enzyme | Methylation Sensitive | Buffer | Water Bath Temperature |
| BamHI | No | 2 | 37 °C |
| EcoRI | Yes | 2 | 37 °C |
| HindIII | No | 2 | 37 °C |
| PmlI | Yes | 1 | 37 °C |
| ScaI | No | 2 | 37 °C |
| SmaI | Yes | 3 | 25 °C |
Table 2: Listed in the table is a subset of possible enzymes that could be used in these experiments. Three enzymes are methylation sensitive and three are not methylation sensitive. The buffers used for the enzymes and incubation temperatures are listed for each enzyme.
