The two methods presented make it possible to follow the kinetic activity and dynamics of protein interactions between a chaperone and its substrate. Moreover, the reduction-oxidation protocol allows the preparation of a fully reduced and fully oxidized chaperone, giving a more in-depth understanding of the activation mechanism of redox-dependent disordered chaperones.
First, we used light scattering in order to examine the redox-dependent activity of the chaperone. Light scattering was performed on a chemically and thermally denatured CS protein in the presence of an oxidized (active) or reduced (inactive) chaperone (Figure 1). Chemical denaturation leads to a rapid CS aggregation induced by the refolding of a fully denaturized protein in unfavorable buffer conditions. On the other hand, the thermal-induced aggregation results from a relatively slow unfolding of the natively folded substrate. Therefore, these different types of aggregation processes vary in their kinetic parameters. Moreover, different chaperones can vary in their ability to prevent the aggregation of the same substrate in different aggregation modes.
In both forms of denaturation, the addition of oxidized Hsp33 in a molar ratio of 1:4 (CS: Hsp33) completely abolished aggregation, lowering the 360 nm readings to negligible values. The presence of a reduced Hsp33, on the other hand, had no effect on the CS stability and gave an aggregation curve similar to the one detected in the absence of a chaperone (Figure 1).The results were analyzed and cleaned from background noise using Kfits, as schematically described in Figure 2.
The outline of the HDX-MS technique begins with the incubation of the chaperone with deuterium oxide, followed by quenching and digestion, and finally MS analysis and hydrogen-deuterium exchange profiling (Figure 3). During HDX-MS, the chaperone was incubated on the tray at 25 °C in a D2O-containing buffer. After a specific incubation time controlled by the sample handling robotic system, the protein solution was transferred to the quenching solution, acidic and denaturing, on the tray at 0 °C. The low pH and low temperature slows the hydrogen exchange and preserves the deuteration levels of all amide hydrogens, while the denaturing chemical unfolds the protein, enabling it to be properly digested. The robotic system transfers the sample from the tray at 0 °C to the cooling box where it flows into the online pepsin digestion column. Immediately after the protein digestion, the peptides proceed to the C18-trap column for the desalting and removal of fast exchangeable D2O (mainly located on the side chain atoms) and are then separated using the C18 column and analyzed using mass spectrometry. The sample flow is controlled by a two-valve system, which 1) ensures the separation of the pepsin and C18 column buffers in order to prevent the inactivation of the pepsin by organic solvents, and 2) enables the short desalting of the peptides to remove fast exchangeable D2O molecules and salts (Figure 4).
After the computational analysis, we receive a deuteration profile for each peptide, showing a change in mass as a function of the incubation time in D2O. The HDX Workbench software allows the comparison of experimental replicates in order to create statistical analyses of each condition (e.g., an active chaperone in an unbound form). The next step is a comparison of the deuteration curves between different samples; for example, a bound chaperone and an unbound chaperone. These comparisons of different chaperone states can reveal the structural perturbations that accompany substrate binding. Residues that exhibit a slower exchange in the bound state are likely interacting with a binding partner. It is important to note that decreased hydrogen-deuterium exchange rates can also indicate a refolding of a specific region upon binding without the actual direct interaction with the substrate (Figure 5A). The HDX Workbench software allows the examination of the results in the "perturbation" view by showing the deuteration with a standard error over the coverage of the whole protein.
In this case, HDX-MS was used to compare the deuteration pattern between the bound and unbound chaperone Hsp33 to its substrate CS. The results reveal a client-interaction site within the C-terminal redox switch domain of the chaperone (Figure 5B). While reduced, Hsp33 has a completely ordered structure, where the interaction sites are buried. Hsp33 loses its structure and becomes functional when it senses oxidative stress; hence, we compared the reduced and oxidized Hsp33 under both bound and unbound conditions (Figure 5C). The results show that while the reduced Hsp33, whether bound or unbound, produces similar peptide curves, the oxidized Hsp33 displays a significant difference. The oxidized Hsp33 samples show a 30% deuteration difference between the bound and unbound chaperone, with specific areas of the bound protein indicating an allosteric hindrance. The analysis of additional peptides from the N-terminal and the thermobile linker regions are shown in Supplementary Figure 3.
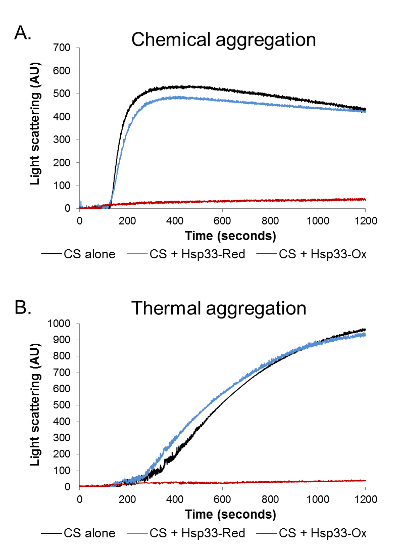
Figure 1. Aggregation analysis by light scattering. The aggregation of chemically and thermally denatured CS was monitored in the presence of oxidized Hsp33 (red), reduced Hsp33 (blue), or in the absence of chaperone (black). A light scattering at 360 nm was monitored for 1,200 s. (A) During the chemical aggregation, in the absence of a chaperone or in the presence of a reduced Hsp33, the CS aggregated rapidly, reaching a plateau approximately 300 s into the measurement. (B) During the thermal aggregation, the CS aggregated gradually in the absence of a chaperone or in the presence of a reduced Hsp33. In both assays, the presence of oxidized Hsp33 completely abolished aggregation, as was demonstrated by negligible 360-nm readings. Please click here to view a larger version of this figure.
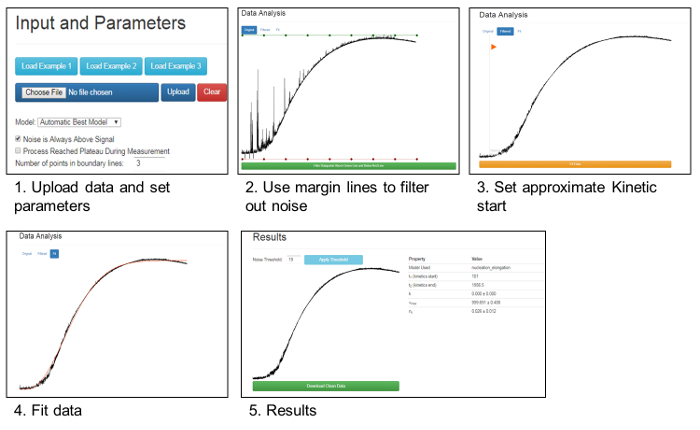
Figure 2. Removal of noise and data analysis using Kfits. This panel shows a schema summarizing the data analysis and noise removal by the Kfits tool, as described in the text and in Rimon et al.45. Please click here to view a larger version of this figure.
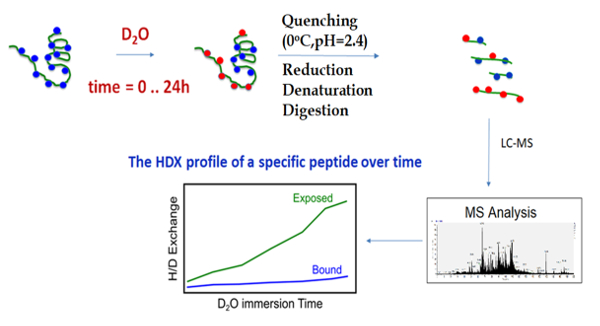
Figure 3. Workflow of the HDX-MS methodology. HDX-MS employs deuterated solvents to determine the chemical environment of amide hydrogens, uniformly distributed along the backbone of the protein. The protein of interest is incubated in a deuterating buffer in its native form for specific time periods, then the hydrogen-deuterium exchange is quenched by acidic conditions at a low temperature. The protein is digested by an enzyme stable under acidic conditions, such as pepsin. Afterward, the peptides are desalted and separated in a short time to avoid a back exchange. Finally, the peptides are analyzed by a mass spectrometer and the deuterium uptake is evaluated by an analysis software, for example by the HDX Workbench free software58. Please click here to view a larger version of this figure.
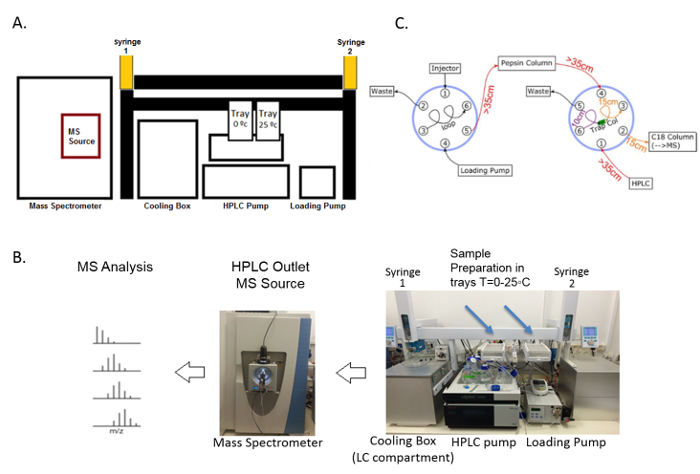
Figure 4. Components of the fully automated HDX-MS system. (A) This schematic diagram of the HDX-MS represents all the parts of the system: the loading pump and the HPLC pump control buffer flow and the sample transfer within the system. The robotic system contains two syringes and trays which are used to prepare the deuterated sample and transfer the proteins to the on-line digestion column via the input valve of the cooling box (LC compartment). Then, the digested peptides are directed to the C18 trap and separation columns, subsequently separated, and directed to the mass spectrometer. (B) This panel shows the actual HDX system setup as it was created in the lab. (C) This panel shows a schematic presentation of the online-digestion and the peptide separation fluidic configuration. The digestion and separation systems have one valve each that can be switched between load and inject modes. The first valve (on the left) is connected to the injection port and is controlled by the loading pump, directing samples into the pepsin column using the acidic buffer without any organic solvent that can damage the pepsin resin. The sample is transferred to the right valve into the trap column to remove residues of deuterium as well as fast exchanged deuterated atoms (mainly of the side chains). After a brief wash of 1 – 2 min in the trap column, the valve changes its mode to load and the sample is washed into the C18 separation column using the HPLC pump with increasing concentrations of acetonitrile. The sample is transferred to the mass spectrometer for a mass analysis. The length of the tubes is marked. Please click here to view a larger version of this figure.
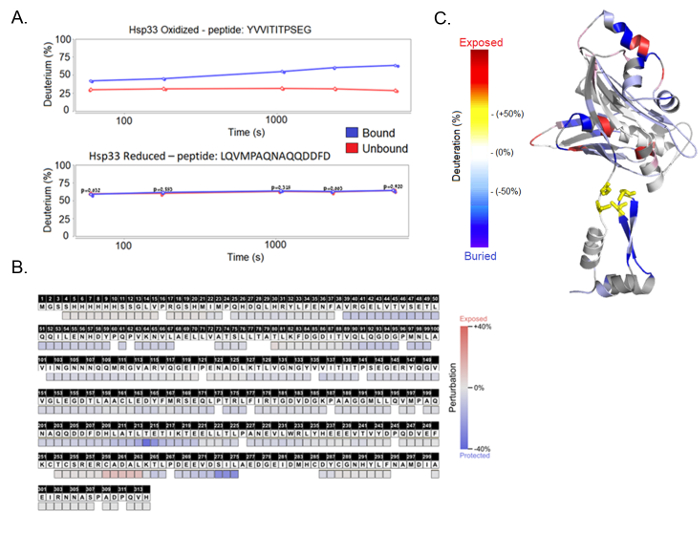
Figure 5. HDX-MS results. (A) The results are presented as a deuteration percentage over time per protein fragment, generated by the HDX Workbench software. Each graph shows a protein sample, either oxidized or reduced, and compares between bound and unbound samples. The peptide fragments shown here are the peptide YVVITITPSEG found in the linker region and the peptide LQVMPAQNAQQDDFD found in the C-terminus. (B) The final results, presented as the deuteration difference between the bound and unbound complexes, show the perturbation per amino acid. (C) A structural model of the Hsp33 chaperone shows the deuteration uptake differences throughout the linker and C-terminal unstable regions. This panel is created using the PyMol software. Please click here to view a larger version of this figure.
Supplementary Figure 1. Interface of the program operating mass spectrometer. The program is used to optimize and define mass spectrometer method parameters used for the HDX experiment. The method parameters (in the left panel) can be saved in the method, such that is recognized by other programs. Please click here to view a larger version of this figure.
Supplementary Figure 2. This figure shows a screenshot of the software which serves as an integration platform of the robotic sampling handing system and the MS-LC system. The software optimizes the running time by the scheduling of the HDX experiment, following the defined incubation time in the deuterated buffer (blue arrows). For example, above is a diagram of 17 runs of two replicative analyses of the Hsp33 variant STIL, incubated in a deuterated buffer for different times, ranging from 0 – 100 min. Please click here to view a larger version of this figure.
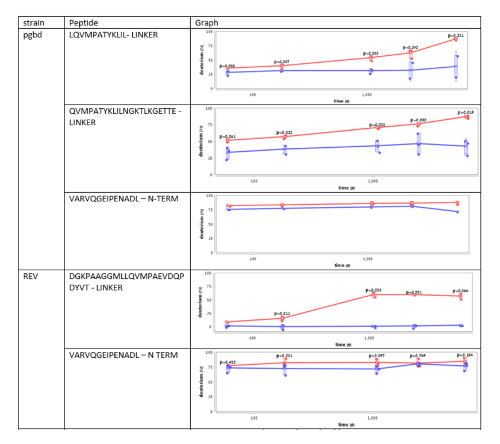
Supplementary Figure 3. This figure shows an analysis of the deuterium uptake of several Hsp33 peptides from different Hsp33 variants, named PGBD and REV34. The plots demonstrate changes in the deuterium uptake of the same region in Hsp33 in unbound (red) and bound (blue) states. Peptides from the N-terminal region show no significant difference in the HDX rates, while fragments from the linker region show a significant decrease in the HDX rates upon the interaction with the unfolded substrate, citrate synthase. Please click here to view a larger version of this figure.
