Identification of Homologous Recombination Events in Mouse Embryonic Stem Cells Using Southern Blotting and Polymerase Chain Reaction
Summary
Here, we present a detailed protocol for identifying homologous recombination events that occurred in mouse embryonic stem cells using Southern blotting and/or PCR. This method is exemplified by the generation of nonmuscle myosin II genetic replacement mouse models using traditional embryonic stem cell-based homologous recombination-mediated targeting technology.
Abstract
Relative to the issues of off-target effects and the difficulty of inserting a long DNA fragment in the application of designer nucleases for genome editing, embryonic stem (ES) cell-based gene-targeting technology does not have these shortcomings and is widely used to modify animal/mouse genome ranging from large deletions/insertions to single nucleotide substitutions. Notably, identifying the relatively few homologous recombination (HR) events necessary to obtain desired ES clones is a key step, which demands accurate and reliable methods. Southern blotting and/or conventional PCR are often utilized for this purpose. Here, we describe the detailed procedures of using those two methods to identify HR events that occurred in mouse ES cells in which the endogenous Myh9 gene is intended to be disrupted and replaced by cDNAs encoding other nonmuscle myosin heavy chain IIs (NMHC IIs). The whole procedure of Southern blotting includes the construction of targeting vector(s), electroporation, drug selection, the expansion and storage of ES cells/clones, the preparation, digestion, and blotting of genomic DNA (gDNA), the hybridization and washing of probe(s), and a final step of autoradiography on the X-ray films. PCR can be performed directly with prepared and diluted gDNA. To obtain ideal results, the probes and restriction enzyme (RE) cutting sites for Southern blotting and the primers for PCR should be carefully planned. Though the execution of Southern blotting is time-consuming and labor-intensive and PCR results have false positives, the correct identification by Southern blotting and the rapid screening by PCR allow the sole or combined application of these methods described in this paper to be widely used and consulted by most labs in the identification of genotypes of ES cells and genetically modified animals.
Introduction
The technology of gene targeting by HR in murine ES cells provides a powerful tool for dissecting the cellular consequences of specific genetic mutations1,2. The importance and significance of this technology are reflected in its recognition by the 2007 Nobel Prize in Physiology or Medicine3,4; meanwhile, it represents the advent of the modern era of gene engineering5. Gene targeting through HR can be utilized to engineer virtually any alteration ranging from point mutations to large chromosomal rearrangements in the genome of mouse ES cells6,7. It is well known that, before the emergence of so-called genome editing tools, the generation of a gene knockout mouse required the application of gene-targeting technology in ES cells8,9,10. During the past two decades, more than 5,000 gene-targeted mice were produced by this approach for modeling human diseases or studying gene functions11. A genome-wide knockout effort has been established for distributing gene-targeting vectors, targeted ES cell clones, and live mice to the scientific community2,12,13,14,15. Undoubtedly, ES cell-based HR-mediated gene-targeting technology has greatly advanced our understanding of the functions of genes played in physiological or pathological context.
Because HR is a relatively infrequent event in mammalian cells16,17, the important and next step following gene targeting in murine ES cells is to analyze numerous ES colonies for identifying a few clones with mutations resulting from HR with the targeting vector18. The gold methods for identifying HR events include Southern blotting and PCR19,20. The advantages of the approaches include that Southern blotting can identify correctly targeted ES clones and allows researchers to analyze the structure of the gene-targeted event, such as a verification of a single copy insertion of the construct, while a PCR-based strategy permits more rapid screening for HR events21,22. Though these methods have drawbacks, such as that they are time-consuming and can have false positives, the combinational usage of them is widely accepted and applied by most labs in identifying HR events, particular in ES cells, for generating genetically modified animals.
Three isoforms of nonmuscle myosin II (NM II) in mammals, each consisting of two identical NMHC IIs which are encoded by three different genes (named Myh9, Myh10, and Myh14) and two pairs of light chains, are referred to as NM II-A, II-B, and II-C23. Previous studies have indicated that at least the isoforms of NM II-A and II-B are essential for mouse development because the in vivo ablation of these isoforms results in embryonic lethality24,25,26. To circumvent this problem and obtain novel insights into the isoform-specific functions of NM II-A and II-B in the later stages of mouse development, a genetic replacement strategy using ES cell-based HR-mediated gene-targeting technology was adopted to generate a series of mouse models27. In the course of identifying the desired ES clones, both Southern blotting and PCR methods were utilized, and these proved to be efficient and reliable27,28.
This paper intends to provide a detailed description of Southern blotting and PCR, including the design of targeting vector(s), probe(s), and primers, and the execution of experiments, as well as the analysis of results exemplified by identifying HR event occurrence in ES cells for creating genetic replacement NM II mouse models and representative data. The protocols of these two methods presented here can also be adopted for identifying the genotypes of genetically modified cells or animals.
Protocol
1. Design of Targeting Construct(s), Probes for Southern Blot, and Primers for PCR
- Select the first coding exon (exon 2) of the Myh9 gene for disruption or insertion in the application of knockout/knock-in reported here.
- Retrieve the 5-kb upstream and 5-kb downstream DNA sequences surrounding the Myh9 exon 2 from the genome.ucsc.edu website.
- Analyze restriction digestion patterns of enzymes (REs) with 1–2 cuts in this 10-kb region by using pDRAW software to determine suitable RE(s) to digest the genomic DNA for Southern blotting.
NOTE: Dra I meets this requirement and is selected for the purpose. - Select a 4 kb fragment immediate upstream of the Myh9 exon 2 as the left homology arm (LHA) and a 1.7 kb fragment immediate downstream sequence as the right homology arm (RHA); choose a 1 kb fragment 5' upstream of the LHA as the left probe (LP) for Southern blotting and a 1.2 kb fragment 3' downstream of the RHA as the right probe (RP), based on the above analysis.
- Use a primer3 program to design the forward and reverse primers for amplifying those four DNA fragments by PCR. Design a pair of primers with the forward primer resided near the 3' terminal of a selection marker neomyocin resistance gene (P1) and the reverse primer located just outside the RHA (P2).
NOTE: This primer pair will be used for identifying targeted ES clones by PCR29. - Find a 129Sv BAC clone covering mouse Myh9 gene locus by visiting the bacpac.chori.org website (note: isogenic DNA is preferred). Isolate BAC DNA using a kit suitable for purifying large pieces of DNA following the instructions provided by the manufacturer.
NOTE: Purified BAC DNA will be used as the template for PCR amplification. - Draw a schematic representation of the targeting constructs, probes, and primers to summarize this information.
2. Generation of Targeting Construct(s) and Probes for Southern Blot, and the Preparation of Primers for PCR
- Order the PCR primers described above and dissolve them into a concentration of 20 µM.
- Amplify the homology arms and probes by PCR in a reaction solution including 1 µL forward and 1 µL reverse primers, 1 µL of BAC DNA (50 ng) as the template, 5 µL of buffer for Pfu and 1 µL of Pfu ultra as the DNA polymerase, and H2O up to 50 µL. Perform the PCR in a PCR machine under the following conditions: 95 °C for 3 min; 95 °C for 30 s; 60 °C for 30 s; 72 °C and 1 kb/min; 30 cycles; finally, 72 °C for 10 min.
- Purify PCR products using a PCR cleanup kit according to the protocol provided by the manufacturer and elute the DNA fragments with 40 µL of H2O. Digest the purified PCR products with predesigned REs in a reaction solution containing 5 µL of 10x buffer for REs, 2.5 µL of RE1 and 2.5 µL of RE2, and the eluted DNA, at 37 °C for 2 h. Run a 1% DNA gel to separate target DNA bands, excise the gel containing the target DNA under the UV light, purify the target DNA fragments from the gel using a DNA gel extraction kit according to the protocol provided by the manufacturer, and elute the DNA fragments with 40 µL of H2O.
- Clone the homology arms and replacement expression cassette(s) into the mpNTKV-LoxP vector according to the order of the amplified and purified RHA, LHA, and replacement expression cassette(s) released from other vectors to obtain the final targeting construct(s). Clone DNA fragments of the amplified and purified probes into the T-easy vector.
- Confirm the nucleotide sequences of all cloned DNA fragments by sequencing27,28.
3. Preparation of Targeting Construct(s), the Electroporation of ES Cells, and the Amplification of ES Clones
- Prepare each targeting construct using a plasmid maxiprep kit according to the protocol provided by the manufacturer. Linearize each construct plasmid by digesting it with Not I in a 400 µL reaction including 40 µL of 10x buffer for Not I, 10 µL of Not I, 100 µg of DNA, and H2O up to 400 µL, at 37 °C overnight.
- Purify the linearized targeting construct(s).
- Extract the digested reaction solution 1x with an equal volume of Phenol:Chloroform:Isoamyl Alcohol (25:24:1) and centrifuge at a force of 2,000 x g for 10 min.
- Transfer the supernatant to a new 1.5-mL tube, precipitate the DNA using 2.5x ethanol and 0.1x 3M sodium acetate (pH 5.2) (volume ratio), and centrifuge at a force of 2,000 x g for 10 min.
- Remove the supernatant, wash the DNA pellet 1x with 1 mL of 75% ethanol, and centrifuge at a force of 2,000 x g for 5 min.
- Remove the supernatant and air-dry the DNA pellet for 5 min.
- Dissolve the linearized DNA pellet in sterile Tris-EDTA (TE) buffer at a final concentration of 1 µg/µL.
- Mix 50 µg of each linearized targeting construct with 0.5 x 107 ES cells. Perform the electroporation at 320 V and 250 µF. Plate the electroporated ES cells onto dishes with neo-resistant MEF feeders.
- After 24 h, switch to ES cell medium with 400 µg/mL G418 and 200 µM ganciclovir and continue to culture for 4–5 days with a daily medium change. Pick up drug-resistant ES clones into 48-well plates.
NOTE: Normally, four 48-well plates are used per construct. - Duplicate the 48-well plates.
NOTE: One set of the plates is cryopreserved, and the other set is used for genomic DNA preparation.
4. Preparation of Genomic DNAs and the Digestion with Restriction Enzyme(s)
- Prepare gDNAs from ES cells using a commercial kit (genomic DNA purification kit) with minor modifications.
- Remove media from the ES cell culture and add 500 µL of nuclei lysis solution, including RNaseA, directly to the wells to lyse the cells.
NOTE: The cell lysate can be stored at -80 °C or treated immediately. - Pipet up and down several times to lyse the cells fully and transfer them to a clean 1.5-mL tube.
- Add one-third of the volume of the protein precipitation solution to the 1.5 mL tube, vortex vigorously for 20 s, chill the samples on ice for 5 min, and then, centrifuge at a force of 2,000 x g for 5 min. Transfer the supernatant to another clean 1.5 mL tube containing an equal volume of isopropanol; gently mix the solution. (Note that white thread-like strands can be seen at this moment.) Centrifuge at a force of 2,000 x g for 1 min; then, discard the supernatant.
- Wash the gDNA pellet with 1 mL of 70% ethanol at room temperature, centrifuge at a force of 2,000 x g for 1 min, aspirate the supernatant carefully, and then, air-dry the gDNA pellet for 3 min.
- Dissolve the gDNAs with 100 µL of DNA rehydration solution and, then, incubate at 65 °C for 1 h or at 4 °C overnight.
- Store the gDNAs at 2–8 °C.
- Remove media from the ES cell culture and add 500 µL of nuclei lysis solution, including RNaseA, directly to the wells to lyse the cells.
- Digest the gDNAs with predesigned RE Dra I. Set up a 30-µL digestion reaction by mixing 3 µL of 10x buffer for Dra I, 3 µL of Dra I, 10 µg of gDNAs/sample, and H2O up to 30 µL, and incubate at 37 °C overnight.
- Check the completeness of the digestion by DNA gel, analyzing 5 µL of the digested reaction, and then, add the 3 µL of 10x DNA-loading buffer for the subsequent step.
5. Southern Blotting and PCR Identification
- Southern blotting screening
- Separate the digested gDNAs by electrophoresis and transfer to a membrane.
- Prepare a 1% agarose electrophoresis gel with ethidium bromide (EB), load the samples from step 4.3 and a 1 kb ladder, and run the gel with a low voltage (30–40 V) overnight.
- Take out the gel and take a picture with a DNA gel-imaging system after electrophoresis. Check whether the digested and separated gDNAs display a smear-like image.
- Soak the gel in a tray with 0.2 N HCl solution and shake it gently for 20 min at room temperature.
- Transfer the gel to DNA-denaturing solution and shake it gently for 20 min at room temperature.
- Switch the gel into DNA-neutralizing solution and shake it gently for 20 min at room temperature.
NOTE: The gel is prone to breakage after this step, so it must be handled carefully. - Use the rapid downward transfer system to transfer the DNAs from the gel to the membrane. Assemble the TurboBlotter and blotting stack according to the instructions provided by the manufacturer.
NOTE: 10x or 20x saline-sodium citrate (SSC) solution is used as a transfer buffer. In general, 3 h of transfer is enough to transfer 95% of gDNAs from gel to membrane; however, a longer time of transfer is innocuous. - Take out the membrane and wash it with 2x SSC for 1 min, absorb the liquid with tissues, and then cross-link the DNA with the membrane using a UV crosslinker.
NOTE: The membrane can be stored at 4 °C for one week.
- Label the DNA probes with radioactivity.
- Purify the probe plasmids using a miniprep kit according to the protocol provided by the manufacturer.
- Release the DNA fragments of the probes from the plasmid vector by EcoR I digestion in a reaction solution including 5 µL of buffer for EcoR I, 2 µL of EcoR I enzyme, 20 µg of plasmid DNA, and H2O up to 50 µL, for 2 h.
- Run a 1% DNA gel for separating the probe DNA fragments from the vector and purify the DNA fragments of the probes with a DNA gel extraction kit according to the protocol provided by the manufacturer.
- Using 1 µL of DNA solution, measure the DNA concentration of the probe DNA fragments with a spectrophotometer at a wavelength of 260/280 nm.
- Prepare 40-ng of probe DNAs in a 1.5 mL tube with 45 µL of TE buffer, boil for 3 min, spin briefly, and then, place the tube(s) on ice for 2 min.
- Add the heat-denatured probe DNAs to the tube containing ready-to-go DNA-labeling beads (-dCTP), pipet up and down to mix, add 5 µL of [α32P]dCTP, and then, incubate at 37 °C for 15 min.
- Purify the labeled probes by using G-50 microcolumns according to the instructions provided by the manufacturer and, then, measure the radioactivity by a scintillation counter (optional).
- Hybridize the membrane(s) with the labeled probes.
- Prehybridize the membrane.
- Prewarm the hybridization solution at 42 °C for 30 min. Mix 20 mL of prewarmed hybridization solution with 200 µg of boiled salmon sperm DNA in a 50-mL tube.
- Place the membrane into the hybridization tube. Add the mixed prehybridization solution to the hybridization tube. Place it into the hybridization oven (set rolling and the temperature at 42 °C) and let the prehybridization proceed for 30 min.
- Hybridize the membrane with the labeled probe(s).
- Take out the hybridization tube and pour the prehybridization solution into a 50 mL tube; add the denatured probe (heated at 100 °C for 3 min) from step 5.1.2.7 to this tube and mix gently.
NOTE: Reduce any inducing bubbles. - Return the mixed solution to the hybridization tube and perform the hybridization at 42 °C overnight.
- Take out the hybridization tube and pour the prehybridization solution into a 50 mL tube; add the denatured probe (heated at 100 °C for 3 min) from step 5.1.2.7 to this tube and mix gently.
- Prehybridize the membrane.
- Wash the membrane(s) to remove nonhybridized probes.
- Place the membrane(s) into a tray with 1x SSC + 0.1% SDS and shake gently at 55–60 °C for 10 min.
- Transfer the membrane(s) to a tray with 0.5x SSC + 0.1% SDS and shake gently at 55–60 °C for 10 min.
- Check the radioactivity on the membrane(s) by using a portable Geiger counter to decide whether a third washing is required.
- Expose the radioactivity on the membrane to X-ray films.
- Remove the liquid from the washed membrane(s).
- Enfold the membrane(s) with plastic wrap and fix it/them in the exposure cassette.
- Expose the membrane to two sheets of X-ray film in a dark room.
- Place the exposure cassette at -80 °C overnight or longer.
- Develop the films to visualize the results. Evaluate whether a corresponding ES clone is the desired one with the targeted recombination or not, according to the sizes of the DNA bands detected by the probes.
- Rehybridize the same membrane by another probe after stripping off the used probe according to the following procedure: take out the used membrane, wash it 1x with clean H2O, and then, incubate it in striping solution (55% formamide, 2% SSPE, 1% SDS, H2O) at 65 °C with gentle shaking for 1–2 h.
- Separate the digested gDNAs by electrophoresis and transfer to a membrane.
- PCR identification
- Perform PCR identification of the desired ES clones in a 50-µL reaction solution including 5 µL of 10x PCR buffer, 2 µL of 50 mM MgSO4, 1 µL of 10 mM dNTP, 1 µL of 20 µM forward primer, 1 µL of 20 µM reverse primer, 1 µL of high-fidelity platinum Taq, gDNAs (~100 ng), and H2O up to 50 µL.
- Use the following PCR reaction conditions: an initial denaturation at 94°C for 3 min, 30 cycles of denaturation at 94°C for 30 s, annealing at 60°C for 30 s and an extension at 68°C for 132 s, and a final step of 68°C for 10 min.
- Analyze the PCR products by electrophoresis in 1.0% agarose.
- Clone the PCR fragments with their expected size into the T-easy vector and sequence to confirm the presence of a partial sequence of the target vector.
Representative Results
In this paper, a detailed protocol of Southern blotting and PCR is described, which is utilized to identify HR events that occurred in mouse ES cells for the generation of NM II genetic replacement mouse models, using ES cells-based HR-mediated targeting technology. Though Southern blotting and PCR, as well as traditional gene-targeting technology, have been widely used for several decades, the successful application of them needs to be planned carefully. At least these aspects are required to be considered: the length of the long and short arms, the positions and length of the probes, the suitable REs for cutting the genomic DNAs, and the primers for PCR, as summarized in Figure 1, which is helpful for subsequent analysis. As an important step of Southern blotting, the prepared and digested genomic DNAs are required to be separated on DNA gel for the detection by the probe. Because genomic DNAs are cut into a lot of fragments with different lengths, they display a smear-like status on the DNA gel, suggesting a complete digestion of the genomic DNAs, as indicated in Figure 2. As a final step of Southern blotting, the signals of a radioactivity-labeled probe hybridizing with a target DNA fragment are shown on the film, which reflect the occurrence of HR events in the ES clones, thereby indicating whether an ES clone is the desired one. According to the predesign in this study, ES clones with mutated allele have two distinct size bands, while wild-type ES clones only have one band, suggesting the desired ES clones are heterozygous (Figure 3). Relative to the procedure and results of Southern blotting, the operation and results of PCR are simple and direct. Following the PCR reaction, the PCR products can be analyzed on the DNA gel. If the PCR bands are specific and sequencing the cloned PCR products confirms the presence of a partial sequence of target vector such as a neo-resistance gene, as well as genomic regions that are just outside of the homology arm, the occurrence of HR events can be expected and verified (Figure 4).
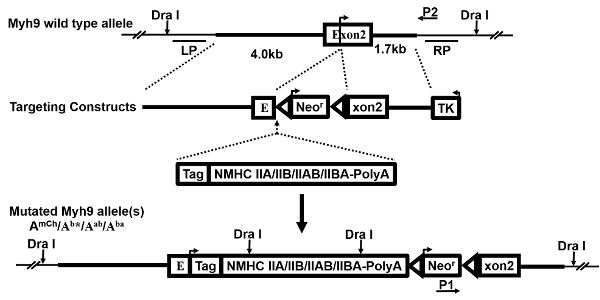
Figure 1: Targeting constructs. This is a schematic demonstrating the generation of multiple targeting constructs. The wild-type (WT) Myh9 gene allele, gene-targeting vector, replacement exogenous expression cassette(s), and the resultant mutated allele(s), as well as the probes (LP, RP) for Southern blot and the primers (P1, P2) for PCR, are shown and described previously27. An arrow on exon 2 indicates the translational initiation site. Following the successful occurrence of HR, the replacement expression cassette and the neomycin resistance gene (Neor) are inserted just 5' of the initiating ATG codon. Therefore, the endogenous Myh9 allele is disrupted and the knocked-in gene(s) is/are expressed in the mutant cells and mice. Please click here to view a larger version of this figure.

Figure 2: Digested genomic DNAs with Dra I. Genomic DNAs from ES clones targeted with the construct replacing NMHC II-A with II-B are digested with Dra I and, then, separated on an agarose gel by electrophoresis. A smear-like digested gDNA is observed. C1 – C8 depict individual ES clones. A complete digestion of gDNA produces a lot of DNA fragments with a different length, thereby displaying a smear-like image. This result also reflects the good quality of prepared gDNAs and the completeness of the digestion. Please click here to view a larger version of this figure.
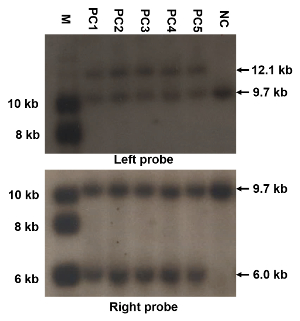
Figure 3: Representative results of Southern blotting. These panels show a Southern blotting screening of the genomic DNAs from ES clones targeted with the construct of replacing NMHC II-A with II-AB, using the left and right probes. The mutated allele shows a 12.1 kb or 6 kb band when the left probe or right probe is used, respectively, while the WT shows a 9.7 kb band. M: marker; PC1-PC5: positive clones; NC: negative clone. The sizes of the Southern blotting bands are also indicated. All procedures of Southern blotting are strictly carried out and the specificity of the probes is good enough; there should be no nonspecific bands expect for the expected bands. Please click here to view a larger version of this figure.
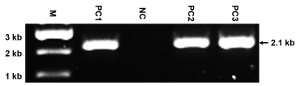
Figure 4: Representative results of PCR. This panel shows the PCR identification of the genomic DNAs from ES clones targeted with the construct of replacing NMHC II-A with II-BA using the primer pair P1 + P2. The mutated allele yields a 2.1 kb band, while the WT allele yields no band. M: marker; PC1-PC3: positive clones; NC: negative clone. The size of the PCR band is also indicated. Since the primers are designed to only detect the mutated allele, the appearance of a single and expected band reflects the specificity of the primers and the high quality of the prepared gDNAs. Please click here to view a larger version of this figure.
Discussion
Currently, designer nucleases for genome editing still cannot replace ES cell-based gene-targeting technology due to its issues of off-target effects, and difficulty in inserting a long DNA fragment30,31. As the golden methods for identifying HR events that occurred in mouse ES cells, this report provides a detailed protocol of Southern blotting and PCR for the field. We validated the reliability of these methods by analyzing individual clones from mouse ES cells targeted with a series of constructs. The desired ES clones identified by these methods had been successfully used to generate corresponding mouse models27.
Though other techniques for the screening of targeted ES clones have been described19,32, the methods of Southern blotting and PCR cannot be completely replaced by those established thereafter32, because these initial techniques have a longer applied history and are widely accepted and confirmed by the scientific society, performed by most biological labs, and are the origin of other technologies. Importantly, the good performance of Southern blotting and PCR in the identification of HR events is well exemplified in previous work29. The results from Southern blotting indicate several unique features: among the randomly screened ES clones, over 90% of them are desired ones, no nonspecific bands are detected, and the HR occurred preferentially on one allele of the Myh9 gene. Meanwhile, the data from PCR, together with sequencing, confirm that the occurrence of HR events is site-specific and match well with those from Southern blotting.
According to our practice, several factors should be considered when Southern blotting and PCR are used to identify HR events in ES cells, thereby obtaining good and expected results. The first one is the length of the homology arms; in general, increasing the homology arm length will enhance the efficiency of HR33. However, this is not always the case. On the one hand, longer arms increase the difficulty of manipulation; on the other hand, the length of the homology arms (4 kb for the left arm and 1.7 kb for the right arm) reported here resulted in the highest HR frequency obtained so far among similar experiments. Additionally, a reasonable length of homology arms facilitates the identification by PCR. The second is the utilization of isogenic DNA for preparing the homology arms and Southern blotting probes34. This can be satisfied by ordering a BAC clone containing the region of the gene-of-interest or by using genomic DNA from the cells intended to be targeted. The third is the selection of suitable REs for digesting genomic DNAs. In general, one RE or the combination of two REs that cut the wild-type or mutant allele only once or twice around the targeting region are preferred; furthermore, the resulting larger DNA fragment should not exceed 15 kb and the size difference between the distinct DNA fragments is over 2 kb. These requirements can facilitate the separation and identification of expected bands by Southern blotting. The fourth is the length of the probes and the least similarity with other sequences in the genome. Generally, the length of the probes is 500–1,000 bp. The similarity with other sequences in the genome can be analyzed with the NCBI BLAST program. Furthermore, a software used to design the probes for Southern blotting has been described35. The fifth factor to be considered is to use the conventional methods to prepare genomic DNA for an enhancing yield. Genomic DNAs prepared from a confluent well of a 48-well plate are generally enough for at least two rounds of Southern blotting analyses. As to designing the primers for PCR, the best strategy is to use one primer present on the selection marker in conjunction with a primer outside of the targeting arms. Additionally, sequencing the PCR products is important for proving HR events20,36. Notably, PCR-based screening cannot completely replace the information obtained through Southern blotting, while it can effectively reduce the numbers of clones to be evaluated.
In conclusion, Southern blotting and PCR are well-demonstrated methods for screening ES clones to identify HR-mediated gene-targeting events in ES cells. Though the detailed protocol described here mainly focused on the screening of desired NM II genetic replacement ES clones, it can be used for genotyping mice that are subsequently generated using the positive ES clones. It can be easily adapted to the identification of HR events in other cell types, such as iPS cells or somatic cells.
Divulgations
The authors have nothing to disclose.
Acknowledgements
This work received support from the General Program of National Natural Science Foundation of China (Grants No. 31571432), the Human Provincial Natural Science Foundation of China (Grant No. 2015JC3097), and the Research Foundation of Education Bureau of Hunan Province, China (Grant No. 15K054).
Materials
| BAC CLONE | BACPAC Resources Center (BPRC) | bMQ-330E21 | |
| QIAGEN Large-Construct Kit | QIAGEN | 12462 | |
| QIAquick Gel Extraction Kit | QIAGEN | 28704 | |
| QIAquick PCR Purification Kit | QIAGEN | 28104 | |
| QIAprep Spin Miniprep Kit | QIAGEN | 27104 | |
| QIAGEN Plasmid Plus Maxi Kit | QIAGEN | 12963 | |
| PfuUltra High-Fidelity DNA Polymerase | Agilent | 600382 | |
| T-easy vector | Promega | A1360 | |
| Nuclei Lysis Solution | Promega | A7941 | |
| Protein Precipitation Solution | Promega | A7951 | |
| DNA Denaturing Solution | VWR | 351-013-131 | |
| DNA Neutralizing Solution | VWR | 351-014-131 | |
| Ready-To-Go DNA Labeling Beads (-dCTP) | VWR | 27-9240-01 | |
| UltraPure SSC, 20X | Thermo Fisher | 15557036 | |
| UltraPur Phenol:Chloroform:Isoamyl Alcohol (25:24:1, v/v) | Thermo Fisher | 15593031 | |
| G418 | Thermo Fisher | 10131035 | |
| Salmon Sperm DNA Solution | Thermo Fisher | 15632011 | |
| Platinu Taq DNA Polymerase High Fidelity | Thermo Fisher | 11304029 | |
| Not I | Thermo Scientific | ER0592 | |
| Dra I | Thermo Scientific | ER0221 | |
| EcoR I | Thermo Scientific | ER0271 | |
| Ganciclovir | Sigma | G2536 | |
| Whatman TurboBlotter Transfer System, Large Kits | Fisher Scientific | 09-301-188 | |
| [α32P]dCTP | PerkinElmer | NEG013H100UC | |
| ProbeQuan G-50 Micro Columns | GE Healthcare | 28-9034-08 | |
| Hybrisol I Hybridization Solution | Millipore | S4040 | |
| Kodak X-Ray Film | Z&Z Medical | 844 5702 |
References
- Gao, G., McMahon, C., Chen, J., Rong, Y. A powerful method combining homologous recombination and site-specific recombination for targeted mutagenesis in Drosophila. Proceedings of the National Academy of Sciences of the United States of America. 105 (37), 13999-14004 (2008).
- Skarnes, W., et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 474 (7351), 337-342 (2011).
- Vogel, G. Nobel Prizes. A knockout award in medicine. Science. 318 (5848), 178-179 (2007).
- Salsman, J., Dellaire, G. Precision genome editing in the CRISPR era. Biochemistry. Cell Biology. 95 (2), 187-201 (2017).
- Capecchi, M. Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nature Reviews Genetics. 6 (6), 507-512 (2005).
- Van, d. W. L., Adams, D. J., Bradley, A. Tools for targeted manipulation of the mouse genome. Physiological Genomics. 11 (3), 133-164 (2002).
- Glaser, S., Anastassiadis, K., Stewart, A. F. Current issues in mouse genome engineering. Nature Genetics. 37 (11), 1187 (2005).
- Bradley, A., Evans, M., Kaufman, M. H., Robertson, E. Formation of germ-line chimaeras from embryo-derived teratocarcinoma cell lines. Nature. 309 (5965), 255-256 (1984).
- Robertson, E., Bradley, A., Kuehn, M., Evans, M. Germ-line transmission of genes introduced into cultured pluripotential cells by retroviral vector. Nature. 323 (6087), 445-448 (1986).
- Thomas, K. R., Capecchi, M. R. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell. 51 (3), 503-512 (1987).
- Skarnes, W. C., et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 474 (7351), 337 (2011).
- Collins, F. S., Rossant, J., Wurst, W. A mouse for all reasons. Cell. 128 (1), 9-13 (2007).
- Poueymirou, W. T., et al. F0 generation mice fully derived from gene-targeted embryonic stem cells allowing immediate phenotypic analyses. Nature Biotechnology. 25 (1), 91-99 (2007).
- Pettitt, S. J., et al. Agouti C57BL/6N embryonic stem cells for mouse genetic resources. Nature Methods. 6 (7), 493-495 (2009).
- Gertsenstein, M., et al. Efficient Generation of Germ Line Transmitting Chimeras from C57BL/6N ES Cells by Aggregation with Outbred Host Embryos. PLoS One. 5 (6), 11260 (2012).
- Smithies, O., Gregg, R. G., Boggs, S. S., Koralewski, M. A., Kucherlapati, R. S. Insertion of DNA sequences into the human chromosomal |[beta]|-globin locus by homologous recombination. Nature. 317 (6034), 230-234 (1985).
- Deng, C., Capecchi, M. R. Reexamination of gene targeting frequency as a function of the extent of homology between the targeting vector and the target locus. Molecular & Cellular Biology. 12 (8), 3365 (1992).
- Lay, J. M., Friishansen, L., Gillespie, P. J., Samuelson, L. C. Rapid confirmation of gene targeting in embryonic stem cells using two long-range PCR techniques. Transgenic Research. 7 (2), 135-140 (1998).
- Langerak, P., Nygren, A. O. H., Schouten, J. P., Jacobs, H. Rapid and quantitative detection of homologous and non-homologous recombination events using three oligonucleotide MLPA. Nucleic Acids Research. 33 (22), 188 (2005).
- Gómezrodríguez, J., et al. Advantages of q-PCR as a method of screening for gene targeting in mammalian cells using conventional and whole BAC-based constructs. Nucleic Acids Research. 36 (18), 117 (2008).
- Kim, H. S., Smithies, O. Recombinant fragment assay for gene targeting based on the polymerase chain reaction. Nucleic Acids Research. 16 (18), 8887-8903 (1988).
- Joyner, A. L., Skarnes, W. C., Rossant, J. Production of a mutation in mouse En-2 gene by homologous recombination in embryonic stem cells. Nature. 338 (6211), 153-156 (1989).
- Ma, X., Adelstein, R. S. The role of vertebrate nonmuscle Myosin II in development and human disease. Bioarchitecture. 4 (3), 88-102 (2014).
- Malonek, D. Relationships between the dynamics of cortical blood flow, oxygenation, and volume changes following sensory stimulation. Proceedings of the National Academy of Sciences of the United States of America. 94, (1997).
- Takeda, K., Kishi, H., Ma, X., Yu, Z. X., Adelstein, R. S. Ablation and mutation of nonmuscle myosin heavy chain II-B results in a defect in cardiac myocyte cytokinesis. Circulation Research. 93 (4), 330-337 (2003).
- Conti, M. A., Evenram, S., Liu, C., Yamada, K. M., Adelstein, R. S. Defects in cell adhesion and the visceral endoderm following ablation of nonmuscle myosin heavy chain II-A in mice. Journal of Biological Chemistry. 279 (40), 41263-41266 (2004).
- Wang, A., et al. Nonmuscle myosin II isoform and domain specificity during early mouse development. Proceedings of the National Academy of Sciences of the United States of America. 107 (33), 14645-14650 (2010).
- Zhang, Y., et al. Mouse models of MYH9-related disease: mutations in nonmuscle myosin II-A. Blood. 119 (1), 238 (2012).
- Liu, T., et al. Identification and characterization of MYH9 locus for high efficient gene knock-in and stable expression in mouse embryonic stem cells. PLoS One. 13 (2), 0192641 (2018).
- Saito, S., Adachi, N. Advances in the Development of Gene-Targeting Vectors to Increase the Efficiency of Genetic Modification. Biological & Pharmaceutical Bulletin. 39 (1), 25-32 (2016).
- Langerak, P., Nygren, A., Schouten, J., Jacobs, H. Rapid and quantitative detection of homologous and non-homologous recombination events using three oligonucleotide MLPA. Nucleic Acids Research. 33 (22), 188 (2005).
- Martin, S. L., et al. A single amino acid substitution in ORF1 dramatically decreases L1 retrotransposition and provides insight into nucleic acid chaperone activity. Nucleic Acids Research. 36 (18), 5845-5854 (2008).
- Kamisugi, Y., Cuming, A. C., Cove, D. J. Parameters determining the efficiency of gene targeting in the moss Physcomitrella patens. Nucleic Acids Research. 33 (19), 173 (2005).
- Luo, Y., Bolund, L., Sørensen, C. B. Pig gene knockout by rAAV-mediated homologous recombination: comparison of BRCA1 gene knockout efficiency in Yucatan and Göttingen fibroblasts with slightly different target sequences. Transgenic Research. 21 (3), 671-676 (2012).
- Croning, M. D., Fricker, D. G., Komiyama, N. H., Grant, S. G. Automated design of genomic Southern blot probes. BMC Genomics. 11 (1), 74 (2010).
- Zimmer, A., Gruss, P. Production of chimaeric mice containing embryonic stem (ES) cells carrying a homoeobox Hox 1.1 allele mutated by homologous recombination. Nature. 338 (6211), 150-153 (1989).
