The recombinant N-terminal nucleoplasmin domain of the protein FKBP53 from Arabidopsis thaliana was subjected to analytical SEC. The elution peak volume was plotted against the standard curve to identify its oligomeric state. The analytical SEC results revealed that the domain exists as a pentamer in solution, with an approximate molecular mass of 58 kDa (Figure 1A,B). Further, the nucleoplasmin domain was analyzed for thermal and chemical stability in conjunction with analytical SEC. The nucleoplasmin sample subjected to heat-treatment up to 90 °C displayed no apparent shift in the elution volume and the peak height compared to the samples maintained at 20 °C, suggesting that the domain is highly thermostable (Figure 1C). Likewise, the nucleoplasmin domain displayed salt stability up to 2 M of NaCl (Figure 1D) and urea stability up to 4 M (Figure 1E). However, the nucleoplasmin pentamer started dissociating in higher urea concentrations.
A pull-down assay was performed to determine the type of interactions contributing to the complex formation between the histone chaperone (nucleoplasmin domain of AtFKBP53) and the histone oligomers H2A/H2B dimer and H3/H4 tetramer using a gradient salt wash. The interaction of the nucleoplasmin domain with H2A/H2B dimer was stable up to a salt concentration of 0.4 M NaCl (Figure 2A). In comparison, the association with H3/H4 was reasonably stable up to 0.7 M NaCl (Figure 2B). The ability of the chaperone-histone complexes to withstand high salt concentration suggests the role of hydrophobic interactions in stabilizing the complexes. The chaperone complex with H3/H4 being stable even in high salt concentrations suggests a predominant role of hydrophobic interactions in the complex formation. The lower stability of the H2A/H2B-chaperone complex in high salt concentrations reveals a significant role for electrostatic interactions in the complex formation. In another experiment, the pull-down assay was used to examine whether the chaperone prefers either H2A/H2B dimer or H3/H4 tetramer. The results revealed that the chaperone binds to H2A/H2B dimer and H3/H4 tetramer simultaneously and irrespective of the order in which they are added to the chaperone (Figure 2C,D). This indicated that the chaperone possesses separate sites for its interaction with the histone oligomers.
AUC-SV experiments (Figure 3) were performed to study the stoichiometry of interaction between histone oligomers and chaperones. AUC-SV data analysis provided a sedimentation coefficient (s) value of 5.40 S for the AtFKBP53 nucleoplasmin domain in complex with H2A/H2B that corresponded to a molecular mass of 104 kDa. The complex of the nucleoplasmin domain with H3/H4 gave a sedimentation coefficient value of 7.35 S, corresponding to 129 kDa. The estimated molecular mass of the complexes reveals that the pentameric nucleoplasmin forms complex with both H2A/H2B dimer and H3/H4 tetramer in a 1:1 stoichiometry.
It is essential to show that the protein can deposit histone oligomers onto DNA to confirm that it is a histone chaperone. Towards this end, a plasmid supercoiling assay was adopted (Figure 4). The relaxed circular plasmid was incubated with the histone oligomers H2A/H2B and H3/H4 with the recombinant plant histone chaperones of the NAP family – AtNRP1 and AtNRP228. The presence of the chaperone increased the amount of supercoiled plasmid, suggesting it could deposit histones onto the DNA to form nucleosomes, causing DNA supercoiling.
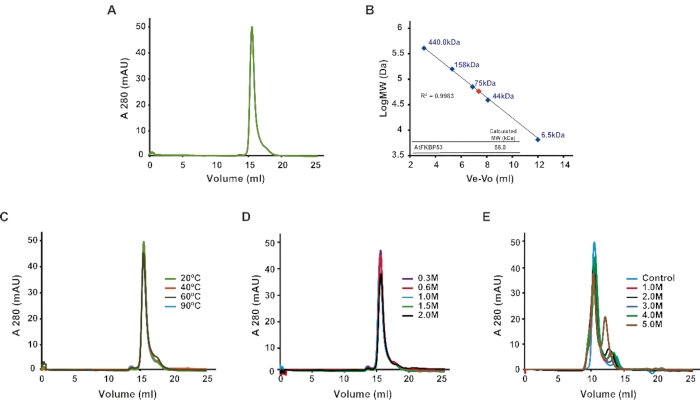
Figure 1: Oligomeric state and stability of the nucleoplasmin domain of AtFBP53. (A) Analytical size-exclusion chromatography profile of the AtFKBP53 nucleoplasmin domain. (B) Calibration curve obtained using globular proteins of known molecular mass. The blue dots represent the molecular mass of the known proteins, whereas the red dot represents the AtFKBP53 nucleoplasmin domain. (440 kDa – ferritin, 158 kDa-aldolase, 75 kDa-con albumin, 44 kDa-ovalbumin, 6.5 kDa-aprotinin). (C) Analytical size-exclusion chromatogram of 500 µL of 0.5 mg/mL AtFKBP53 nucleoplasmin domain subjected to heat treatment at different temperatures: 20 °C (green), 40 °C (orange), 60 °C (black), 90 °C (light blue). (D) Analytical size-exclusion chromatogram of 500 µL of 0.5 mg/mL AtFKBP53 nucleoplasmin domain in buffers containing different NaCl concentrations: 0.3 M (purple), 0.6 M (red), 1.0 M (light blue), 1.5 M (green), 2.0 M (black). (E) Analytical size-exclusion chromatogram of the AtFKBP53 nucleoplasmin domain in buffers with different urea concentrations: 0 M (control; light blue), 1.0 M (pink), 2.0 M (black), 3.0 M (dark blue), 4.0 M (green), 5.0 M (brown). The nucleoplasmin pentamer shows high stability to thermal and chemical stress conditions. The figure is adapted from Reference21. Please click here to view a larger version of this figure.
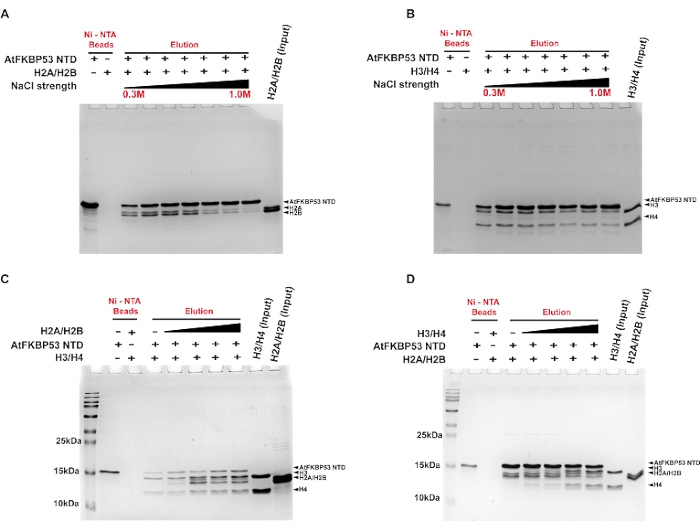
Figure 2: Pull-down assays for the interaction of the nucleoplasmin domain of AtFKBP53 with histone oligomers. 18% SDS-PAGE images of the elution fractions from the assays are presented here. Pull-down assay for (A) 20 µM H2A/H2B dimer and (B) 20 µM H3/H4 tetramer with 5 µM AtFKBP53 nucleoplasmin domain in increasing concentrations of NaCl in the range of 0.3 M, 0.5 M, 0.6 M, 0.7 M, 0.8 M, 0.9 M, and 1.0 M. 5 µM AtFKBP53 FKBD was used as a negative control here. For the competitive binding experiment, (C) a mixture of 5 µM AtFKBP53 nucleoplasmin domain and 20 µM H3/H4 tetramer incubated with a range of 20-60 µM H2A/H2B dimer and (D) a mixture of 5 µM AtFKBP53 nucleoplasmin domain and 20 µM H2A/H2B dimer incubated with a range of 20-60 µM H3/H4 tetramer has been used. The label AtFKBP53 corresponds to the nucleoplasmin domain of AtFKBP53. Elution fractions show simultaneous binding of both the histone oligomers to the nucleoplasmin. The figure is adapted from Reference21. Please click here to view a larger version of this figure.
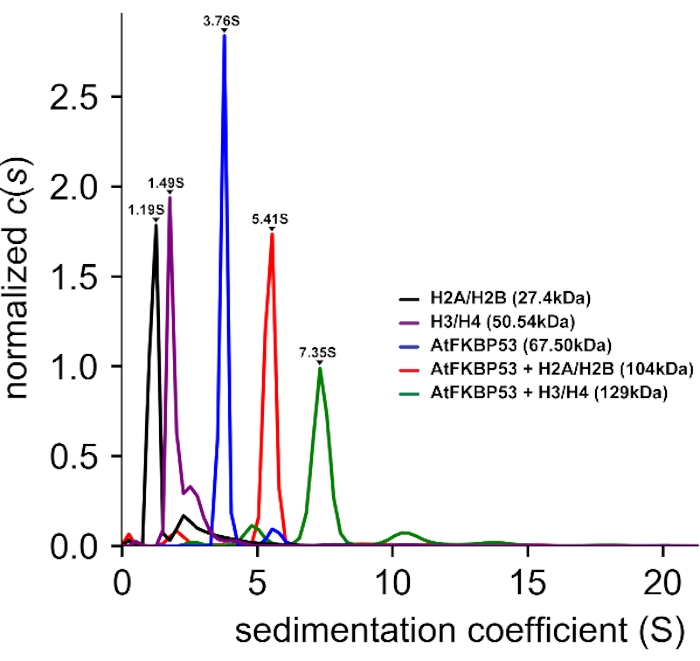
Figure 3: Analytical ultracentrifugation – sedimentation velocity (AUC-SV) experiment of histone oligomers, the nucleoplasmin domain of AtFKBP53, and their complexes. The AUC distance distribution vs. sedimentation coefficient (S) plot. The obtained sedimentation coefficient (s) values and molecular masses are also provided. The label AtFKBP53 corresponds to the nucleoplasmin domain of AtFKBP53. The estimated molecular masses reveal a 1:1 stoichiometry for the AtFKBP53 nucleoplasmin domain with the histone oligomers H2A/H2B dimer and H3/H4 tetramer. 450 µL of all the protein samples having an OD280 of 0.3-0.5 were used for the AUC-SV experiments. The figure is adapted from Reference21. Please click here to view a larger version of this figure.
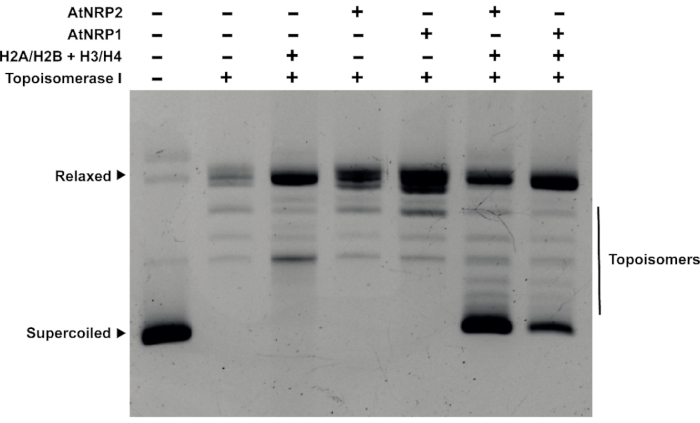
Figure 4: Plasmid supercoiling assay. Plasmid supercoiling assay for the histone chaperones AtNRP1 and AtNRP2. 500 ng of pUC19 plasmid DNA was pretreated with 1 µg of Topoisomerase I for the experiment. 4 µM AtNRP1, 4 µM AtNRP2, and a mixture of 4 µM H2A/H2B dimer and 2 µM H3/H4 tetramer were as control that shows no supercoiling activity when incubated with the pretreated pUC19 DNA. The lanes with a mixture of 4 µM H2A/H2B of dimer and 2 µM H3/H4 of tetramer and 4 µM each of AtNRP1 and AtNRP2 show the formation of supercoiled DNA upon incubation with the pretreated pUC19 DNA. Please click here to view a larger version of this figure.
