Immunostaining and flow cytometry characterization of iPSC-derived CMs, ECs, and CFs
To generate cardiac microtissues composed of iPSC-CMs, iPSC-ECs, and iPSC-CFs, all three cell types are differentiated and characterized individually. In vitro differentiation of iPSCs to iPSC-CMs has improved over the past several years. However, the yield and purity of iPSC-CMs differ from line to line. The current protocol yields over 75% pure iPSC-CMs that spontaneously start beating around day 9 (Figure 1A). Further purification steps from day 9 to day 14 can improve iPSC-CM purity to over 80% as previously described12. Similarly, high-purity iPSC-ECs can be generated using previously published protocols13,14 that include the addition of several vascular growth factors that polarize endothelial progenitors arising from the mesoderm around days 4-5 (Figure 1B) to form phenotypically well-defined iPSC-ECs. iPSC-CFs are a highly heterogenous population based on their location and are characterized based on their morphology and expression of extracellular matrix proteins. Here, using published protocols15,16,17 with modifications, human iPSC-CFs are obtained from cardiac mesoderm progenitor cells (Figure 1C).
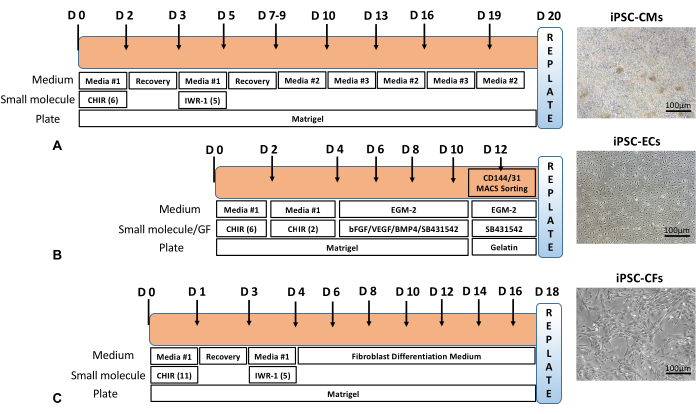
Figure 1: Differentiation timeline. Overall schematic of (A) iPSC-CM, (B) iPSC-EC, and (C) iPSC-CF differentiation timeline with representative phase contrast images of cells after differentiation and purification steps. Scale bar = 100 µm. Please click here to view a larger version of this figure.
The purity of iPSC-CMs, iPSC-ECs, and iPSC-CFs were determined by immunostaining and flow cytometry using cTnT2, platelet endothelial cell adhesion molecule (PECAM1/CD31) and vimentin (VIM), respectively (Figure 2A). Quantitative analysis showed a purified population containing over 90% cTnT2 cells at day 20. iPSC-ECs obtained at day 12 after MACS using CD31 beads were identified with immunostaining against PECAM1/CD31 endothelial cell surface marker. MACS yielded highly pure endothelial cells as evidenced by over 95% CD31+ cells at P0. However, it must be noted that the purity of endothelial cells decreased with higher passage numbers due to de-differentiation. Similarly, at day 20, flow cytometry analyses revealed that over 95% iPSC-CFs expressed the fibroblast marker VIM (Figure 2B).
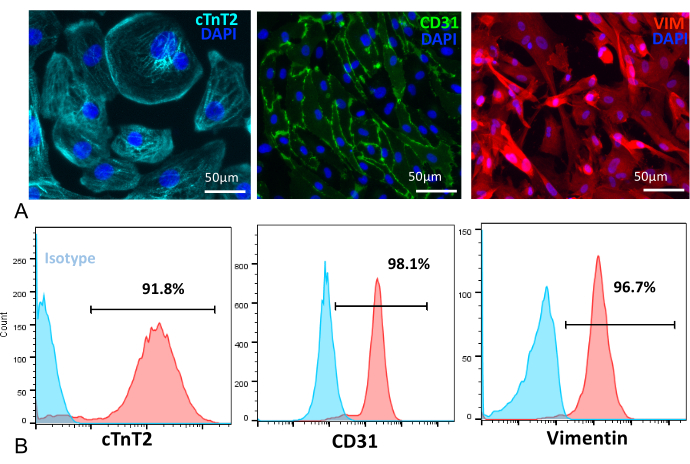
Figure 2: Immunofluorescence and flow cytometry characterization. (A) [Left to right panel] iPSC-CMs at day 25 stained with cTnT2 (cyan), iPSC-ECs stained with PECAM1/CD31 (green), iPSC-CFs stained with VIM (red), and nuclei stained with DAPI (blue). Scale bar = 50 µm. (B) [Left to right panel] Flow cytometry quantification showed a high percent purity of iPSC-CMs (91.8%), iPSC-ECs (98.1%), and iPSC-CFs (96.7%) following differentiation. Please click here to view a larger version of this figure.
Fabrication of cardiac microtissue cultures and size analyses
Single cell suspensions of iPSC-CMs, iPSC-ECs, and iPSC-CFs were mixed in 7:2:1 ratio and carefully dispensed into the cell seeding chamber of the sterilized agarose replica mold (Figure 3A,B). The cells uniformly settled inside the circular recesses in 2 h. Around day 3, the self-assembled cells organize into uniform sized spontaneously beating cardiac microtissues (Figure 3C). Arrays of different sized microtissues can be fabricated by tuning the final cell density (Figure 3D,E). Cardiac microtissues fabricated with a final cell density of 1 x 106 cells/mL is ~300-350 µm in diameter, 2 x 106 cells/mL is ~600 µm in diameter, and 4 x 106 cells/mL is over 800 µm in diameter. Microtissue assembly was obtained with a cell density of 1 x 106 cells/mL, which is typically used for experiments. These microtissue cultures can be maintained in culture for up to 6 weeks.

Figure 3: Replica molding technique to generate multicellular cardiac microtissues. (A) Replica molded agarose microwell trays of (B) iPSC-CM, iPSC-EC, and iPSC-CF mixtures captured inside the microwells for self-assembly. Scale bar 500 = µm. (C) Micrograph showing compaction of cardiac microtissues on day 3. Scale bar = 500 µm. (D) Cardiac microtissue sizes formed show a linear relationship with initial seeding densities, with higher cell densities resulting in larger microtissues. (E) A representative figure showing the self-assembled cardiac microtissues in the microwell array. Please click here to view a larger version of this figure.
Immunostaining and viability after enzymatic digestion
Immunofluorescence staining of day 12 post-fabrication using antibodies against cTnT2 for iPSC-CMs, CD31 for iPSC-ECs, and DDR2 for iPSC-CFs revealed a unique cell distribution in cardiac microtissues. iPSC-CMs, the heaviest of all three cell types, occupied the center, whereas iPSC-ECs were interspersed throughout the microtissues, and iPSC-CFs were observed to predominantly occupy the periphery (Figure 4A). Short and rapid digestion of the microtissues achieved using Dispase I and Liberase TL resulted in overall highly viable cell proportion (Figure 4B, top panel) with less than 5% apoptotic cells after 2 weeks in culture. This was followed by a brief 1 h exposure of cardiac microtissues to a high concentration (5 µM) of Doxorubicin, a chemotherapeutic drug that is known to induce dose-dependent cardiotoxicity (Figure 4B, bottom panel).
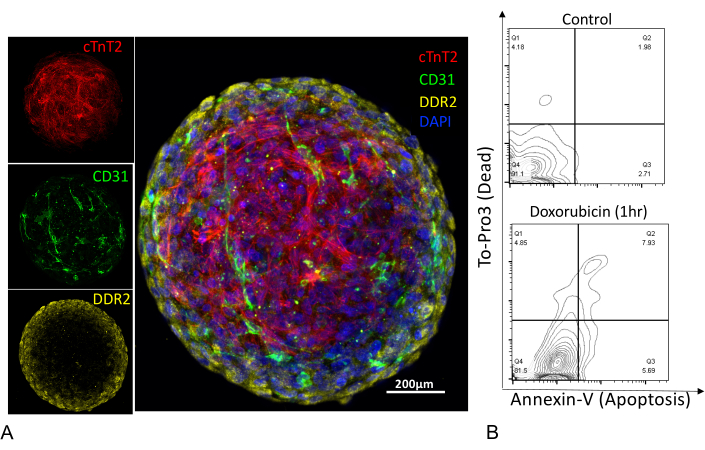
Figure 4: Immunostaining of cardiac microtissues and assessment of cell viability. (A) Confocal z-stack images of human cardiac microtissue stained for iPSC-CMs (cTnT2), iPSC-ECs (CD31), iPSC-CFs (DDR2), and nuclei stained with (DAPI). Scale bar = 200 µm. (B) Flow cytometry plots of cardiac microtissues enzymatically digested into single cell suspension and stained with Annexin V (apoptotic marker) and dead cell exclusion dye (To-Pro3). Digested single cell suspension showed a high cell viability (91%) with <5% apoptotic cell population (top panel), compared to single cell suspension treated with an apoptosis-inducing chemotherapeutic drug, Doxorubicin, for 1 h at 5 µM concentration (bottom panel). Please click here to view a larger version of this figure.
Computational contractility analysis
Contractility analyses of individual cardiac microtissues can be performed with the help of a MATLAB-based image analyses tool. Video recordings of spontaneously beating cardiac microtissues were obtained at 30 fps for analysis. As described previously11, the block-matching method employs motion tracking algorithm to capture movement of a block of pixels for the total frames acquired as a time series of motion vectors. The contractility of the microtissues and movement of the vectors generate a pseudo heatmap that illustrates mean or average contraction profile across the microtissue (Figure 5A). Contractile motion of the cardiac microtissues generates positive peaks that are measured as contraction velocity (blue circle), relaxation velocity (red triangle), and beat rate, the last of which is calculated as the time between two contraction cycles (Figure 5B). Furthermore, the contractility of the cardiac microtissues do not change significantly over 4 weeks in culture (Figure 5C).
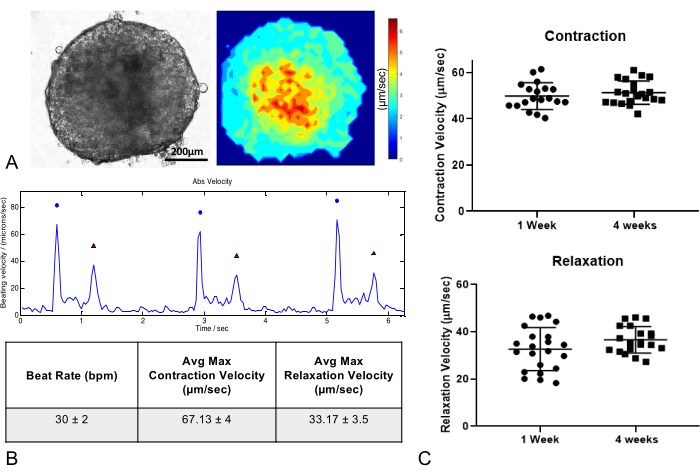
Figure 5: Contraction analysis of cardiac microtissues. (A) Phase contrast image and contraction map of cardiac microtissues 1 week after fabrication. Scale bar = 200 µm. (B) Cardiac microtissues show regular contraction and relaxation profiles and beat rates. Table shows representative values of beat rate, maximum contraction, and relaxation velocities. (C) Long-term culture of cardiac microtissues for up to 4 weeks does not significantly influence contractility parameters (n = 20/group). Please click here to view a larger version of this figure.
