


Overview
ソース: ドミニク・R・ボリノ1, エリック・A・レゲンゾフ2, トーニャ・J・ウェッブ1
1メリーランド大学医学部微生物学・免疫学科、マーリーン・スチュワート・グリーンバウム総合癌センター、ボルチモア、メリーランド州 21201
2メリーランド大学医学部生体医工学技術センター、ボルチモア、メリーランド州 21201
共焦点蛍光顕微鏡は、従来の「広視野」上蛍光顕微鏡と比較して光学分解能の向上を可能にするイメージング技術である。共焦点顕微鏡は、一度に試料の各ピクセルにレーザー照明を指示する一連の電圧制御ミラー(ガルバノメーターまたは「ガルボミラー」)を使用して、x-y光学分解能の向上を達成することができます。さらに重要なのは、ピンホールを使用して、スキャンするZ面にない場所から発信されるフォーカス光を除去することで、優れたZ軸分解能を実現し、検出器が指定されたZ平面からデータを収集できるようにすることです。共焦点顕微鏡ではZ解像度が高いため、一連のZプレーン(Zスタックとも呼ばれる)から画像を収集し、ソフトウェアを介して3D画像を構築することが可能です。
共焦点顕微鏡のメカニズムを議論する前に、サンプルが光とどのように相互作用するかを考慮することが重要です。光は、光子、電磁エネルギーのパケットで構成されています。生物学的サンプルに衝突するフォトンは、4つの方法のいずれかでサンプルを含む分子と相互作用することができます:1)フォトンは相互作用せず、サンプルを通過します。2)フォトンが反射/散乱されます。3)光子は分子によって吸収され、吸収されたエネルギーは、総称して非放射性崩壊として知られているプロセスを通じて熱として放出されます。そして4)光子が吸収され、エネルギーが蛍光と呼ばれるプロセスを通じて二次光子として急速に放出される。蛍光発光を可能にする構造を持つ分子を蛍光素子と呼んでいます。ほとんどの生物学的サンプルには、ごくわずかな内因性蛍束蛍が含まれています。したがって、外因性蛍光体は、サンプル内の関心のある特徴を強調するために使用する必要があります。蛍光顕微鏡検査の間、サンプルは蛍光色素による吸収のための適切な波長の光で照らされる。光子を吸収すると、蛍光素は「励起」と言われ、吸収の過程は「励起」と呼ばれます。蛍光色素が光子の形でエネルギーを放出すると、プロセスは「発光」と呼ばれ、放出された光子は蛍光と呼ばれます。
蛍光色素を励起するために使用される光線は、顕微鏡の対物レンズによって焦点を合わせ、最大に焦点を合わせている「焦点」に収束します。焦点を超えて、光は再び発散します。入り口と出るビームは、焦点に触れる円錐のペアとして視覚化できます(図 1、左パネルを参照)。回折の現象は、光線をどれだけしっかりと集めることができるかに制限を課します - ビームは実際に有限サイズのスポットに焦点を合わせます。焦点点の大きさを決定する2つの要因:1)光の波長、および2)対物レンズの集光能力は、その数値絞り(NA)によって特徴付けされる。焦点「スポット」は、x-y平面だけでなく、z方向にも広がり、実際には焦点ボリュームです。この焦点容積の次元は光学イメージ投射によって達成可能な最高の決断を定義する。フォトンの数は焦点ボリューム内で最も多くなりますが、焦点の上下の円錐形の光パスには、フォトンの密度も低くなります。光路の蛍光色素は、このように興奮することができます。従来の(広視野)上蛍光顕微鏡では、焦点面の上下の蛍光が焦点外蛍光(「かすんだ背景」)に寄与し、画像の解像度とコントラストを低下させます。焦点面(赤い星)の上の蛍光を表す赤い立方体は、焦点の外の蛍光(右上)をもたらします。この問題は、ピンホールの利用のために、共焦点顕微鏡で改善される。(図2、右下)を参照してください。図3に示すように、ピンホールは、焦点の場所から検出器(左)に到達するために発生する放出を可能にし、焦点外の蛍光(右)が検出器に到達するのを妨げ、したがって、解像度とコントラストの両方を向上させます。
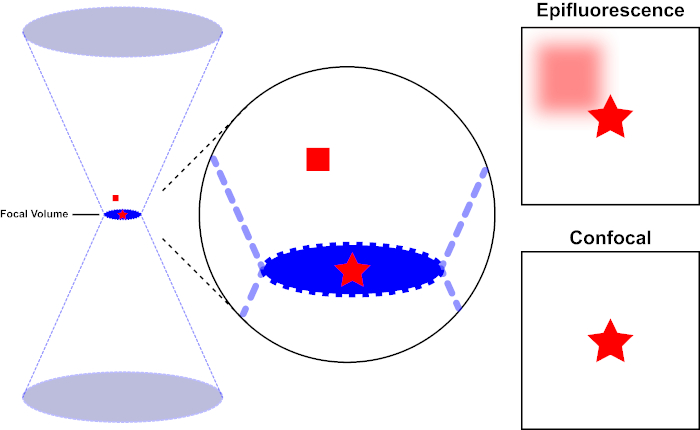
図 1.エピ蛍光と共焦点顕微鏡の光学分解能。この図のより大きなバージョンを表示するには、ここをクリックしてください。
蛍光色素を励起するために使用される光線は、顕微鏡の対物レンズによって焦点を合わせ、焦点量で収束し、発散する(左)。赤い星はイメージされているサンプルの焦点面を表し、赤い正方形は焦点面の上の蛍光色素放出を表します。エピ蛍光顕微鏡を使用してこのサンプルの画像を撮影すると、焦点が合っていない赤い正方形からの放出が見え、「曇った背景」(右上)に寄与します。共焦点顕微鏡は、焦点面の外側に放出される光の検出を防止するピンホールを備えており、「曇った背景」(右下)を排除します。

図 2.共焦点顕微鏡におけるピンホール効果この図のより大きなバージョンを表示するには、ここをクリックしてください。
励起光の最高強度はレンズの焦点(左、赤楕円形)にありますが、焦点にないサンプルの他の部分(右、赤い星)は光と蛍光を得ます。これらの焦点外領域から放出される光が検出器に到達するのを防ぐために、ピンホールを持つ画面が検出器の前に存在する。焦点面から放射されるインフォーカスライト(左)のみがピンホールを通過し、検出器に到達できます。ピンホールでピンホールでピンホールでピンホールがブロックされ、検出器に到達できないピンフォーカスライト(右)。
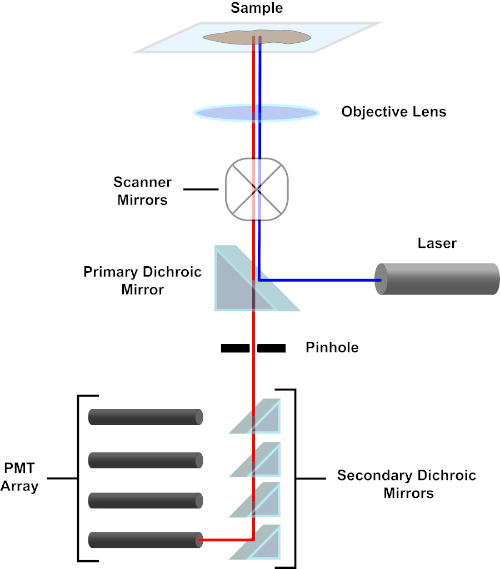
図 3.共焦点レーザー走査顕微鏡の主成分。この図のより大きなバージョンを表示するには、ここをクリックしてください。
簡潔にするために、共焦点顕微鏡の機械的記述はニコンエクリプスTi A1Rの機械的記述に限定されます。異なる共焦点顕微鏡間にわずかな技術的な違いがあるかもしれませんが、A1Rは共焦点顕微鏡機能を記述するための良いモデルとして役立ちます。ダイオードレーザーの配列によって生成される励起光線は、画像化される標本に光を集中させる目的に一次ダイクロイックミラーによって反射される。一次ダイクロイックミラーは、他の波長の光が通過できるようにしながら、励起光を選択的に反射します。次に、光は、X-yの方法で標本全体の光ビームをスイープする走査ミラーに遭遇し、一度に 1 つの(x,y)ピクセルを照らします。照らされたピクセルで蛍光が放出される蛍光は、対物レンズによって収集され、一次二色鏡を通過して光増倍管(PMT)の配列に到達する。二次二次二色ミラーは、放出光を適切なPMTに向ける。ライト パスと PMT に到達します(図 3 を参照)。これにより、比較的弱い蛍光を励起光線から散乱する光によって汚染されることなく定量化することができ、これは通常、蛍光よりも大きさの大きさである。ピンホールは焦点ボリュームの外側から光をブロックするため、検出器に到着する光は、選択された狭いZ面から来ます。したがって、画像は一連の隣接するz-平面から収集できます。この一連の画像は、多くの場合「Z スタック」と呼ばれます。適切なソフトウェアを使用することにより、z-stackを処理して、試料の3D画像を生成することができます。共焦点顕微鏡検査の特に利点は、染色の細胞内局在を区別する能力である。例えば、細胞内染色からの膜染色との分化は、従来のエピ蛍光顕微鏡(1,2,3)では非常に困難である。
サンプル調製は、共焦点イメージングの重要なファセットです。光学顕微鏡検査技術の強みは、生細胞または固定細胞を画像化する柔軟性です。3D画像を生成しようとすると、Zスタックのために取得する必要がある画像の数、細胞の健康維持の難しさ、および生細胞とその小器官の動きのために、固定細胞の使用が典型的です。共焦点蛍光のための細胞を固定および染色するための手順は、免疫蛍光において従来使用されているものと同様である。チャンバースライドまたはカバースリップで培養した後、細胞は細胞形態を維持するためにパラホルムアルデヒドを使用して固定される。非特異的抗体結合は、ウシ血清アルブミン、ミルク、または正常血清を用いて遮断される。二次抗体の特異性を維持するために、使用される溶液は、一次抗体が生成されたのと同じ種に由来してはならない。細胞は、目的の抗原を結合する一次抗体でインキュベートされる。複数の細胞標的を標識する場合、一次抗体はそれぞれ異なる種から誘導されなければならない。抗原をタグ付けする抗体は、フッ素結合二次抗体によって結合される。蛍光結合二次抗体は、共焦点顕微鏡で利用可能なレーザー励起の波長と互換性があるように選択する必要があります。複数の抗原を可視化する場合、蛍光体の励起/放出スペクトルは、その信号が顕微鏡分析によって判別できるように十分に異なる必要があります。染色された標本はイメージ投射のためのスライドに取付けられる。取り付け媒体は、光漂白や試料の脱水を防ぐために使用されます。必要に応じて、核カウンターステインを含む取り付け媒体(例えばDAPIまたはホエヒト)を使用することができる(4)。
以下のプロトコルでは、CD1d(LCD1)を発現するためにトランスフェクトされたマウス線維芽細胞を、CD1dおよびCD107a(LAMP-1)を認識する抗体で染色した。CD1dは、脂質抗原を提示する抗原提示細胞の表面に存在する主要組織適合性複合体1(MHC1)様受容体である。LAMP-1(リソソーム関連膜タンパク質-1)は、主にリソソーム膜に存在する膜タンパク質である。適切な抗原の提示のために、CD1dは低pHリソソームコンパートメントを通して入稿されるので、LAMP-1はこのプロトコルのリソソームコンパートメントのマーカーとして使用されている。異なる種で産生された抗CD1dおよび抗LAMP-1を用いてLCD1細胞を調べることによって、ユニークな蛍動管を持つ二次抗体を使用して、細胞内の各タンパク質の局在化およびCD1dがLAMP-1陽性に存在するかどうかを決定することができる。リソソームコンパートメント。
Procedure
1. 材料
バッファー
- 洗浄バッファー:カルシウムまたはマグネシウムを使用せずに1 X無菌リン酸緩衝生理食塩分(PBS)を使用
- 固定バッファー: PBS における 1% パラホルムアルデヒド
- 透過性バッファー: 0.1% トリトン X-100 (PBS)
- ブロッキングバッファー: PBSにおける1%ウシ血清アルブミン
- 細胞増殖培地:10%の胎児ウシ血清(FBS)、ペニシリン/ストレプトマイシン、およびL-グルタミンを補充したDMEM
機器
- ラミナーフローフード
- 加湿インキュベーター (37°C, 5% CO2)
- 共焦点レーザー走査顕微鏡;ここでは、ニコンエクリプスTiレーザー
材料・試薬
- チャンバリード細胞培養スライド
- DAPIを用いるアンチフェード取り付けメディア(核染色用)
- 顕微鏡カバーガラス
- デリケートなタスクワイプ
- ピペッタとヒント
アッセイ特異的試薬
- 付着細胞(一次細胞または細胞株);。ここで、CD1dをトランスフェクトしたマウス線維芽細胞を用いた(LCD1)。
- 細胞標的を検出する一次抗体;ここで、ラットアンチマウスCD107a(LAMP-1)およびマウス対マウスCD1dを用いた。
- 一次抗体アイソタイプに特異的なフルオロフォア結合二次抗体;ここで、アレクサフルオール488に結合した抗ラットIgGと、Alexafluor 647に結合した抗マウスIgGが使用された。
2. プロトコル
抗体染色の準備
シードセル
- 成長培地に関心のあるセルを再中断します。
- 次に、細胞懸濁液/1当たりのシード500μLを4ウェルチャンバスライドのウェルに入れた。(ここで、LCD1細胞を2.5x105細胞/チャンバで500μLの増殖培養培養物に播種した。播種密度は、細胞株によって異なる場合があります)。
- 37°Cで5%のCO2インキュベーターで一晩チャンバースライドをインキュベートし、細胞がガラスに付着できるようにします。
- 翌日、各井戸から培った培体を吸引し、500 μL PBSで細胞1Xを洗浄します。
固定
- 細胞を固定するには、各ウェルに500 μL 1%パラホルムアルデヒド溶液を加え、室温で15分間インキュベートします。
- インキュベーション後、パラホルムアルデヒドを適切な有害な液体廃棄物容器に回収する。
- 次に、500 μL PBSで細胞を3回洗浄し、固定剤の残骸を除去する。
パーミア化
- 500 μL透過性バッファー/ウェルで室温で15分間インキュベートして細胞を透過化します。
- 次に、500 μLのPBSで細胞を3回短時間洗浄します。
ブロック
- 4°Cで1時間500μLブロッキングバッファーを用いて各ウェルの細胞をインキュベートし、非特異的抗体結合を遮断する。
一次抗体インキュベーション
- スライドチャンバからブロッキングバッファを吸引します。
- 次いで、希釈した一次抗体溶液の500μLを細胞に添加する。(ここで、抗CD107a(LAMP-1)を1:500希釈し、抗CD1dを未希釈して使用した(1H6モノクローナル抗体はランディ・ブルトキエヴィッチ博士によって親切に提供された)。
- スライドを4°Cで一晩インキュベートします。
注: 複数の標的を調べている場合は、一次抗体が異なるアイソタイプであることを確認してください。推奨抗体濃度はメーカーによって異なり、使用前に定量する必要があります。
二次抗体インキュベーション
- 一次抗体溶液をウェルから吸引する。
- 500 μL PBSで井戸室を4回洗います。
- 次に、希釈二次抗体溶液の500μLを各ウェルに添加する。(ここで、二次抗体-抗マウスIgG Alexafluor 647および抗ラットIgG Alexafluor 488のいずれも、ブロッキングバッファー内で1:2000を希釈した。
- 暗闇の中で1時間室温でインキュベートする。
- インキュベーション後、二次抗体溶液を吸引する。
- 500 μL PBSでチャンバーを4回洗浄し、非結合二次抗体を除去します。
注: 推奨抗体濃度はメーカーによって異なるため、使用前に計算する必要があります。複数の標的を調べた場合、二次抗体は、独特の励起/発光スペクトルを持つ異なる蛍光色素に結合する必要があります。また、蛍光色素を選択しながら、核カウンターステイン(すなわちDAPI)の励起/放出を心に留めておいてください。蛍光色素選択は、使用される共焦点顕微鏡のレーザー構成によって影響を受ける可能性がある。機械のレーザー構成は実験に適した蛍光色素を決定する。
3. 取り付けカバースリップ
- まず、慎重にスライドからチャンバーを取り外します。
- 次に、繊細なタスクワイプの上にスライドを斜めに保持し、セルに触れることなくエッジから流体を取り除きます。
- 核染色DAPIを含むアンチフェード取り付け培地を1滴、細胞の各セクションに加えます。
- 次に、指先を使用して端に保持して、スライドの上に20 mm x 60 mmのカバースリップを配置します。(細胞の上に泡が形成され、イメージングを妨げるため避けてください)。
- 繊細なタスクワイプで側面の余分な取り付け媒体を拭き取り、1週間まで室温で暗闇の中でスライドを保存します。
4. 共焦点イメージング
共焦点レーザー走査顕微鏡上の画像サンプル。図2に示すデータについては、ニコンエクリプスTi A1RをNISエレメンツ先端研究ソフトウェアで使用しました。次のセクションでは、前述のソフトウェアを使用してイメージをキャプチャする手順について詳しく説明します。
- 細胞のイメージングを開始するには、'NISソフトウェアアイコン'をクリックして'NIS要素高度な研究ソフトウェア'を開きます。
- 次に、コントロールウィンドウで「TiPad」タブをクリックし、イメージングの目的を選択します。(ここでは、最初の40倍の目的が使用されました)。
- ステージにセルを積み込み、レンズの下に中央にスライドをロードします。
- さて、「A1plusコンパクトGUI」タブで、使用する蛍光色素に適したレーザーを設定します。ギアシンボルをクリックして色素とスペクトル設定メニューを開き、必要なチャンネルを選択し、各チャンネルのレーザーを設定します。
- 次に、最初のダイクロイックミラーの下にあるドロップダウンメニューで適切な排出量を選択します。
- 次に、「A1plusコンパクトGUI」ウィンドウの下で、'Ch.シリーズ'をクリックしてラインチャンネルシリーズを設定し、使用するレーザーがサンプルに同時に発射されるか、順番に発射するかを設定します。(ここでは、チャネル 1 から始まり、チャネル 2、次に 4 から順次パスを選択しました)。
- その後、上部の矢印の先端アイコンをクリックしてスキャンを開始します。この時点で、イメージングがライブ中に'A1plus Compact GUI'ウィンドウの下で、スライドスケールをクリックし、ピンホールサイズを変更してピンホールサイズを変更して、ピンホールサイズを変更して、フォーカスライトの切り下げを保証します。(ここでは、使用可能な最小設定 (0.5) が使用されました)。
- 次に、スライドスケールを使用して、潜在的な背景染色を制限しながら特定の染色を検出できるようにすることで、各レーザーの「高電圧」および「オフセット」設定を適切なレベルに調整します。正染色サンプルが利用可能な場合は、レーザー設定がノイズ比に最適な信号を得られるように、各チャンネルに対してこのサンプルをイメージングから開始します。
注意:長期間のレーザー強度が高いと、光漂白の原因となります。 - 各レーザーに最適なHVとオフセット値を設定した後、[ND取得]タブ'をクリックし、Zシリーズのパラメータを設定する'Z'アイコンを選択します。次に、サンプルのライブ画像を取得しながら、まず画像の下部を見つけて'下部'ボタンをクリックして下部を設定し、次にサンプルの上の位置を見つけ、'上'ボタンをクリックします。ステップ サイズを設定するには、各ステップごとに優先ステップ サイズを μm で入力するか、必要な合計ステップ数を指定します。
- Z シリーズ パラメータを設定したら、画像の必要なサイズ/ピクセル解像度を選択します。これを行うには、'A1plusコンパクトGUI'ウィンドウをクリックし、'サイズ'アイコンの下に目的の解像度を選択します。画像のノイズを減らすには、'ø'記号の横にあるドロップダウンメニューを選択して、選択した画像数を平均化します。
- サンプルのイメージングを開始するには、[ND取得] メニューの [今すぐ実行]タブをクリックします。
- イメージングが完了したら、[ファイル'をクリックしてイメージを保存し、次に 'として保存'をクリックして、拡張子.nd2' を持つイメージ ファイルをエクスポートします。最後に、他の各サンプルのプロセスを繰り返します。
共焦点蛍光顕微鏡は、抗体共役蛍光色素で抗原を標識し、蛍光シグナルを検出することにより、細胞または組織サンプルに関心のあるタンパク質または抗原の局在化のための特殊なイメージング技術です。広視野蛍光顕微鏡よりも高い空間分解能を提供し、対物レンズの焦点面に配置された2つのピンホールの助けを借りて、その名前を共焦点に与えます。細胞内染色からの表面膜染色の分化など、細胞内レベルで染色を可視化できます。
共焦点顕微鏡は、古典的な蛍光顕微鏡と同様の基本原理に従います。光源からのビーム(通常は共焦点用レーザー)は、ダイクロイックミラーによって反射され、サンプル上の対物レンズによって焦点を当てます。この光は、カメラやアイピースに対して対物レンズとダイクロイックミラーを通って戻って移動する異なる波長を放出するために蛍光を励ます。
共焦点顕微鏡の高められた決断は主に励起および放出の光の道を通る光のための非常に小さい穴である2つのピンホールの存在によるものである。ピンホールは、対物レンズの焦点面に戦略的に配置されます。次に、顕微鏡配置の側面図回路図に切り替えて、光路を確認してみましょう。励起ピンホールを通過した後、励起光ビームは焦点から発信する効果を有し、対物レンズがサンプル上の点にも光を集中させます。この焦点からの放出ビームは、通過することを可能にする発光ピンホールに収束します。今、励起の間に、光経路内の蛍光体は、焦点の上下に、わずかに興奮している。焦点から発生する発光光がピンホールを通過する間、焦点外のポイントからの放出は、放出ピンホールの前後に収束し、したがってブロックされ、バックグラウンド蛍光が減少します。
励起放出検出サイクルは、いくつかの異なる方法で行うことができる、対象領域の各イメージングポイントに対して繰り返される必要があります。例えば、レーザー走査共焦点は、異なる角度で励起光をそらすガルバノメーター走査ミラーを使用します。したがって、XY平面内の試料を横切って光線を掃引する。スピニングディスクコンフォーカルは、ピンホールの配列を持つディスクを使用し、ピンホールの配置をシフトするために回転します。これにより、ユーザーはサンプル内の複数の小さなイメージングポイントを毎回照らし、ディスクが回転するにつれて領域全体を徐々にカバーできます。ピンホールの結果として、検出器のXY画像は狭いZ平面を表します。したがって、画像は、多くの場合、Z スタックと呼ばれる一連の連続した Z 平面から収集できます。これらの画像から、適切なソフトウェアは、サンプル中の蛍光シグナルパターンの3D描写を生成することができる。
このプロトコルでは、マウス線維芽細胞の免疫染色を観察し、続いて共焦点顕微鏡でイメージングを行い、細胞表面タンパク質とリソソームタンパク質を可視化します。
始めめるために、無菌技術を使用して、ウェル当たり500マイクロリットルの成長培地に目的の細胞を再懸濁し、次に4ウェルチャンバースライドの井戸にそれらを播種する。ここでは、抗原提示分子CD1dを発現するためにトランスフェクトしたマウス線維芽細胞を用いている。細胞がガラスに付着できるように、チャンバースライドを37°Cの5%の二酸化炭素インキュベーターに入れ、一晩インキュベートします。午前中に、各井戸から培布を吸引し、その後、数秒間PBSの500マイクロリットルで一度細胞を洗浄します。
細胞を固定するには、各ウェルに1%のパラホルムアルデヒド溶液の500マイクロリットルを追加し、室温で15分間インキュベートします。インキュベーションの後、適切な有害な液体廃棄物容器にパラホルムアルデヒドを収集し、数秒間PBSで細胞を3回洗浄することにより、固定剤の残骸を除去します。
細胞への抗体の浸透を可能にするために、各ウェルに500マイクロリットルの透過性バッファーを追加し、室温で15分間ベンチでインキュベートする。パーメビリゼーション後、500マイクロリットルのPBSで細胞を3回短時間洗浄します。次に、各ウェルに500マイクロリットルのブロッキングバッファーを追加し、非特異的抗体結合を防ぐために摂氏4度で1時間インキュベートする。
一次抗体、抗CD1dおよび抗LAMP-1を、適切な作業濃度で調製する。次いで、ウェルからバッファーを吸引し、希釈された一次抗体溶液の500マイクロリットルで各ウェルの細胞を覆い、その後、4°Cで平らな表面に一晩スライドをインキュベートします。翌朝、二次抗体を希釈し、この場合、明確な蛍光タグを有する抗マウスおよび抗ラット抗体を、適切な作業濃度に対する遮断バッファー内で。次に、一次抗体溶液をウェルから吸引し、500マイクロリットルのPBSで細胞を4回洗浄する。次いで、希釈した二次抗体溶液の500マイクロリットルを各ウェルに添加し、暗闇の中で室温で1時間インキュベートする。インキュベーション後、二次抗体溶液を吸引し、500マイクロリットルのPBSでウェルを4回洗浄し、結合のない二次抗体を除去する。
最終洗浄後にサンプルを取り付けるには、慎重に取り外し、スライドからチャンバーを取り外します。残留PBSを除去するには、繊細なタスクワイプの上にスライドを持ち、セルに触れることなくエッジから流体を取り除きます。余分なPBSが除去されたら、核染色DAPIを含むアンチフェード取り付け媒体を1滴、細胞の各セクションに加えます。次に、20×60ミリメートルのカバースリップを取り、指先だけを使用して、細胞上の気泡の形成を避けるために、いずれかのエッジでゆっくりとカバースリップを下げ始めます。デリケートなタスクワイプでスライド上の余分な取り付け媒体を拭き取り、1週間まで室温で暗い状態でスライドを保管します。
セルのイメージングを開始するには、まずデスクトップの NIS ソフトウェア アイコンをクリックします。コントロールウィンドウで上部のTiPadタブをクリックし、画像化の目的を選択します。次に、スライドをセルをステージにロードし、レンズの下に中央に置きます。次に、TiPad タブの横にある A1plus コンパクト GUI タブで、使用する蛍光色素に適したレーザーを設定します。歯車記号をクリックして、色素とスペクトル設定メニューを開きます。色素とスペクトル設定メニューが開いたら、必要なチャンネルを選択し、各チャンネルにレーザーを設定します。次に、最初のダイクロイックミラーの下にあるドロップダウンメニューで適切な排出量を選択します。次に、A1plusコンパクトGUIウィンドウの下で、Ch.Seriesをクリックしてラインチャンネルシリーズを設定し、使用するレーザーがサンプルに同時に発射されるか、順番に発射するかを設定します。
その後、上部の矢印先端アイコンをクリックしてスキャンを開始します。この時点で、イメージングがライブである間、A1plus Compact GUI ウィンドウの下で、スライドスケールをクリックし、ピンホールサイズを変更してピンホールサイズを変更して、ピンホールサイズを変更して、焦点外の光を制限します。次に、スライドスケールを使用して各レーザーの高電圧とオフセットの設定を適切なレベルに調整し、潜在的な背景染色を制限しながら特定の染色を検出できるようにします。正の染色サンプルが利用可能な場合は、レーザー設定が最適な信号対雑音比を得られるように、各チャンネルに対してこのサンプルをイメージングから開始します。各レーザーに最適なHVとオフセット値を設定した後、ND取得タブをクリックし、Zアイコンを選択してZシリーズのパラメータを設定します。
次に、サンプルのライブ画像を取得しながら、まず画像の下部を見つけて下ボタンをクリックして下部を設定します。次に、サンプルの一番上の位置を見つけ、上部ボタンをクリックします。ステップ サイズを、各ステップごとに優先ステップ サイズをミクロンで入力するか、必要な合計ステップ数を指定して、ステップ サイズを設定します。画像の目的のサイズ/ピクセル解像度を選択するには、[Aiplus コンパクト GUI] ウィンドウをクリックし、サイズアイコンの下で目的の解像度を選択します。
画像のノイズを減らすには、theta シンボルの横にあるドロップダウン メニューを選択して、選択した画像数を平均化します。この後、ND 取得メニューの [今すぐ実行] タブをクリックして、サンプルのイメージングを開始します。イメージングが完了したら、ファイルをクリックしてイメージを保存し、次のように保存し、拡張子 dot-nd2 でイメージ ファイルをエクスポートします。最後に、他の各サンプルのプロセスを繰り返します。
この実験では、表面糖タンパク質遺伝子CD1dを発現するマウス線維芽細胞を固定し、免疫染色し、共焦点顕微鏡上で画像化した。この画像は、CD1dが赤で染色された40倍の倍率でZスタックの単一のセクションを示しています。試料を、リソソームマーカーであるLAMP-1を緑色で受け取った。核染色DAPIは、細胞の核を示すために使用された。
3 つの異なるチャネルがマージされる複合イメージでは、赤チャネルと緑チャネルの重なりから黄色の外観が生じ、CD1d と LAMP-1 がリソソームで共局在する領域を示します。1 色のみ存在する領域は、共ローカライズなしで CD1d または LAMP-1 が存在することを示します。この画像は、Z スタックでキャプチャされたイメージから構築されたセルの 3D レンダリングを示しており、このメソッドを使用すると、このセル グループのサイド ビューの構築が可能になります。次の図は、100倍の倍率でZスタックからスライスを示し、これら2つのタンパク質の発現パターンをより詳細に示しています。イメージの右側にあるピンクの輪郭を描いたボックスには、イメージ内のピンクの線で指定された X 座標の断面が表示され、ピンクの線の側面図が表示されます。同様に、イメージの下部にある青い輪郭を描いたボックスは、青い線の正面図を表す青い線で指定された y 座標の断面を示しています。Z スタック イメージの 3D レンダリングにより、ユーザーは 3D でイメージを表示し、X、y、および z 平面をすべて視覚化できます。これは、細胞内の異なる領域における異なる汚れの共局在を研究するために使用することができる。
Subscription Required. Please recommend JoVE to your librarian.
Results
この実験では、表面糖タンパク質遺伝子CD1dを発現するマウス線維芽細胞を固定し、共焦点顕微鏡上で免疫染色し、画像化した。上記のプロトコルを用いて得られた代表的な画像を図4に示す。Aの上部パネルには、個々の標的の染色パターンを示す単一チャネル画像が提示される。これらのイメージは、キャプチャされた Z スタックの単一のセクション (スライス) で構成されます。右のパネルは、細胞の核のDAPI染色を示しています。中央パネルには、CD1dが赤色に染色され、リソソームマーカーであるLAMP-1が緑色で染色されています。左側のパネルは、3 つの異なるチャネルがマージされる合成イメージです。赤と緑のチャネルの重なりから黄色の外観が発生し、CD1d と LAMP-1 が共にローカライズされている領域を示します。染色の結果は、CD1dがLAMP-1+エンドソームコンパートメントに局在していることを確認する。また、CD1d または LAMP-1 が共ローカライズされていない場合に、1 色しか存在しない領域もあります。A の下部パネルには、Z スタックでキャプチャされたイメージから構築されたセルの 3D レンダリングが表示されます。
パネルBは、これらの2つのタンパク質の発現パターンをより詳細に示す100倍倍のZスタックのスライスを示す。イメージの右側にあるピンクの輪郭を描いたボックスには、イメージ内のピンクの線で指定された X 座標の断面が表示され、ピンクの線の側面図が表示されます。同様に、イメージの下部にある青い輪郭を描いたボックスには、青い線の正面図を表す青い線で指定された y 座標の断面が表示されます。Z スタック イメージの 3D レンダリングにより、ユーザーは 3D でイメージを表示し、すべての x、y、Z 平面を視覚化できます。

図4:CD1d及びLAMP1の染色。この図のより大きなバージョンを表示するには、ここをクリックしてください。
A、トップパネル:LCD1細胞を固定し、透過化し、CD1d(赤色)およびLAMP-1(緑、リソソームコンパートメントのマーカー)に対する抗体で染色した。DAPI(青色、核を可視化するために用いられた)。マージ(左パネル)は、CD1dがLAMP-1陽性後期エンドソーム/リソソームコンパートメント(黄色)に局在していることを示しています。
A、下部パネル:トップパネルで同じセルの3Dレンダリング。画像は、NISエレメント先端研究ソフトウェアを使用して、ニコンエクリプスTiの40倍の油浸し目的を使用して取得されました。
B:画像の下部に特定のy座標(青い線で示される)のスタック情報を含む、Aのように染色されたLCD1dセルの100倍の画像(青いボックス)。特定の X 座標 (ピンクの線で示される) のスタック情報が、イメージの右側 (ピンクのボックス) に表示されます。
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
共焦点蛍光染色は比較的単純な手順であり、従来の蛍光顕微鏡と同様の方法で調製された標本の非常に高品質な画像をもたらす。簡単に言えば、サンプルは固定され、透過化され、その後ブロックされる。目的のタンパク質またはタンパク質に対する一次抗体は結合を可能にし、次に蛍色素共役二次抗体が染色を可視化するために使用される。共焦点蛍光顕微鏡は、研究の多くの分野で応用を有する。例えば、目的のタンパク質と共に細胞下小器官のマーカーを染色することにより、共焦点顕微鏡を使用して、多様なタンパク質の細胞内位を決定することができる。従来の蛍光顕微鏡と比較して、共焦点イメージングは、タンパク質の細胞表面と細胞内位置をより効果的に区別することができます。さらに、共焦点イメージングを使用して、2つのタンパク質が細胞内で共局所化するかどうかを決定することもできます。このプロトコルでは概説されていないが、共焦点蛍光顕微鏡はまた、動的変化を検出するために生細胞上で行うことができる。

ビデオ 1:NISエレメント高度な研究ソフトウェアで作成されたビデオは、画像の3Dレンダリングを移動する機能を強調しています。このビデオを見るにはここをクリックしてください(右クリックしてダウンロードしてください)。
Subscription Required. Please recommend JoVE to your librarian.
References
- Claxton, N. S., Fellers, T. J. and Davidson, M. W. Laser scanning confocal microscopy. Department of Optical Microscopy and Digital Imaging, National High Magnetic Field Laboratory, Florida State University, 37 p., Unpublished (2010). Available at- http://www.vertilon.com/pdf/PP6207.pdf.
- Ojcius, D. M., Niedergang, F., Subtil, A., Hellio, R. and Dautry-Varsat, A. Immunology and the confocal microscope. Research in Immunology, 147 (3),175-88 (1996).
- Paddock, S. W. and Eliceiri K. W. Laser scanning confocal microscopy: history, applications, and related optical sectioning techniques. Methods in Molecular Biology, 1075, 9-47 (2014).
- Hoff. F. How to prepare your specimen for immunofluorescence microscopy. Philipps University Marburg, Institute of Cytobiology and Cytopathology, Germany. (2015) Available at- http://www.leica-microsystems.com.