1. Animal Perfusion5 and Whole Mouse Brain Clearing
- The entire length of the procedure can vary depending on the length of time used per dehydration step, but in total the whole process can be conducted in two days.
- Weigh YFP mice and then deeply anesthetize with an intraperitoneal injection of ketamine/xylazine (100 mg/kg:10 mg/kg).
- Confirm a surgical plane of deep anesthesia before proceeding to surgery. Check the animal every 5 min to see if it reacts to a firm toe or tail pinch. If the animal reacts, a supplemental dose (1/3 of original dose) of ketamine/xylazine is required.
- Once deeply anesthetized, restrain the mouse by adhering each limb to a surgical bed using lab tape so that the mouse is in a supine position (dorsal recumbancy), exposing its chest for surgery. The surgical bed is usually made of a metal or plastic mesh and placed into a sink or on top of a lipped-pan so that blood and fixative waste can easily be collected.
- To begin, make an incision below the xyphoid process. Cut along the base of the rib cage using scissors and tweezers and pull back skin as the cut is made. Make two cuts along either side of the mouse sternum (through the ribs) to create a flap of tissue that is held away from the chest cavity using a hemostat to leave the heart exposed.
- Insert a 23 g needle into the left ventricle of the heart and make a small incision in the muscle wall of the right atrium to allow blood to escape. Please refer to JoVE article 2497 for a video of this procedure.5
- Immediately after the right atrium is cut, begin a perfusion with 4 °C phosphate buffered saline, PBS, (pH 7.2) until blood is no longer observed draining from the right atrium of the heart (30 – 40 ml at a rate of about 5 ml/min).
- Once all the blood has been drained (fluid exiting the right atrium is clear), switch the perfusion medium to a chilled 4% PFA solution. Perfuse until the mouse’s body becomes noticeably rigid and cold to the touch (approx. 30 – 40 ml at a rate of about 5 ml/min). (Concentrated 16% PFA solution is diluted according to manufacturer’s instructions and NaOH is added until pH 7.2 is reached. Wear gloves and a lab coat and handle and mix chemicals inside a fume hood.)
- After perfusion, remove the mouse from the surgical bed and decapitate to begin the excision of the brain.
- Using forceps and iris scissors, remove the skull in small sections starting from the back of the skull and moving forward. Make small cuts every 2 – 4 mm with the scissors across the skull while using the forceps to carefully pull the bone away from the brain in small sections. Do this until the entire top surface of the brain is exposed.
- Excise the brain from the skull using a 5 mm wide, flat spatula and place into a glass vial. Submerge in 4% PFA for 6 hr at 4 °C for post fixation.
- After post fixation, wash the brain twice in room temperature PBS by pouring the PFA out of the glass vial and replacing it with PBS. Swirl the brain in PBS solution before pouring out and replacing with PBS for a second washing.
- Dehydrate the brain at room temperature by a graded series of ethanol incubations (once in 50%, 70%, 95%, and twice in 100%) at 2 hr per incubation, and then 12 hr for the second 100% ethanol incubation, to extract the water from the fixed tissue. For each incubation, pour out the previous ethanol solution from the glass vial and replace it with the subsequent solution until the brain is completely submerged.
- After the second incubation in 100% ethanol, pour out the solution and replace with equal parts ethanol and the clearing solution containing benzyl alcohol and benzyl benzoate (1:2 vol:vol ratio). After 2 hr of incubation, decant this solution and replace with a 100% solution of benzyl alcohol and benzyl benzoate (1:2 vol:vol ratio). The refractive index of the clearing solution is n = 1.54.
- Once in BABB, the brain will become noticeably transparent, within 4 – 5 hr. For best clearing results, leave the brain to clear for 6 days at room temperature while shielded from bright light.
2. Microscope Setup
- Once the brain is cleared and ready for imaging, affix it to the bottom of a Petri dish using cyanoacrylate. Allow the adhesive to dry before proceeding.
- After drying, submerge the brain in BABB and place the Petri dish under the microscope objective for imaging.
- We use a multiphoton microscope that incorporates a Mai Tai titanium sapphire laser adjustable between a 710 nm to 990 nm excitation wavelength. The excitation wavelength we use to generate YFP signals is 886 nm. Laser power varies from 30 – 100 mW depending on imaging depth.
- Capture the reflected fluorescent signal using a Nikon 5X objective (NA, 0.5) that allows for large field-of-view imaging (2 x 2 mm).
- Filter the reflected fluorescent signal using a 535/50 bandpass filter and collect it using a GaAsP PMT (H7422PA-40, Hamamatsu, Bridgewater, New Jersey).
- Process images using ScanImage software6 at a resolution of 2048 x 2048 pixels using a scan rate of 2 ms per line to generate high-resolution, YFP images.
- Once imaging is complete, remove the brain from the Petri dish using forceps and store in BABB shielded from light for future imaging. For glued samples, remove the tissue by running a razor blade between the glue and the Petri dish at an angle no greater than 30 degrees. We have stored samples in BABB for up to 1 year with no visible degradation.
3. Representative Results
The representative images and videos shown here demonstrate the high-resolution multiphoton imaging capability made possible by optical clearing. Whole brain imaging allows for YFP-labeled neurons in the different layers of the hippocampus and cortex to be clearly visible as deep as 2 mm below the tissue surface. Figure 1 shows a representative image of a coronal view corresponding to the anatomical features of the hippocampus at 2.92 mm caudal to bregma7. The depth of imaging in the sample was 1.94 mm. Some of this difference was due to removal of the cerebellum at the caudal end of the brain and the balance was due to shrinkage from the dehydration process. Another image from the stack shows layer V/VI pyramidal neurons of the neocortex expressing YFP 2 mm beneath the tissue surface (Figure 2). All images were acquired using the Nikon 5X objective and magnification was done using the digital zoom feature for image acquisition in ScanImage software.
By zooming in on the neocortex, the individual axons and neuron cell bodies of pyramidal neurons in Layer V of the neocortex are clearly distinguishable up to 1.02 mm beneath the tissue surface (Figure 3). Using the image stack from Figure 3, a 3D reconstruction of the neuron region was made using ImageJ software8 (Figure 4).
The high-resolution capability of MPM also allows us to present images of whole, individual neurons. Here we show a reconstruction of a Layer V pyramidal neuron of the neocortex in which individual dendritic processes are clearly visible (Figure 5).
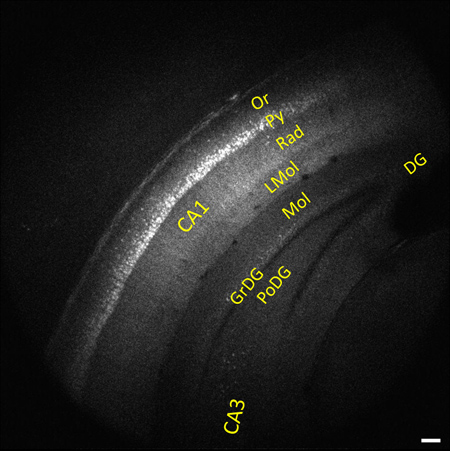
Figure 1. Representative image from a 1.2 mm image stack (0.8 – 2 mm beneath the tissue surface) of whole mouse brain showing neocortex and the different layers of the hippocampus. Features of the hippocampus are visible 1.1 mm below the tissue surface and have been labeled using the following code: DG = dentate gyrus, GrDG = granular layer of DG, Lmol = lacunosum moleculare layer, Mol = molecular layer DG, Or = oriens layer, PoDG = polymorph layer DG, Py = pyramidal cell layer, Rad = stratum radiatum.7 Image is 1.8 x 1.8 mm in size, scale bar = 200 μm. The rostral end of the brain was affixed to a Petri dish with the caudal end facing up toward the objective. This facilitated imaging in the coronal plane.
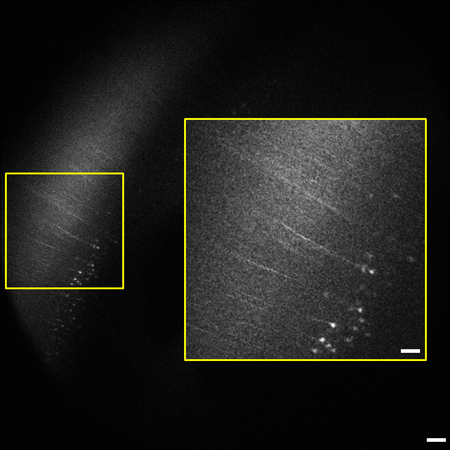
Figure 2. Representative frame from the 1.2 mm image stack (0.8 – 2 mm beneath the tissue surface) of whole mouse brain highlighting the neocortex. Pyramidal cells and processes expressing YFP in layer V/VI of cortex are visible 2 mm below the tissue surface. The degree of labeling is consistent with previous reports of the sparse labeling in the neocortex.9 Inset (right) is enlarged from boxed area on left. Image is 1.8 x 1.8 mm in size, scale bar = 200 μm (50 μm inset).
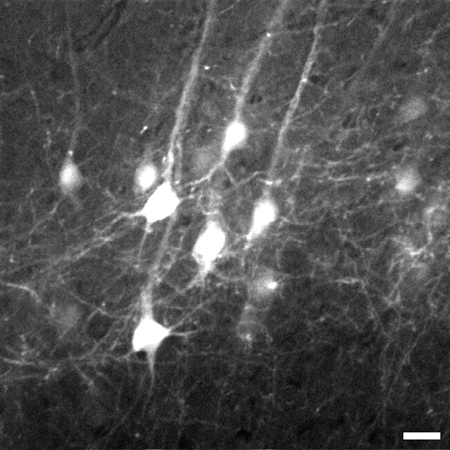
Figure 3. Representative frame taken 774 μm beneath the tissue surface from the image stack of layer V pyramidal neurons in the neocortex of the brain. Stack goes from 700 μm to 1,020 μm beneath the tissue surface. An 8X digital zoom was used to capture details such as fine processes. Image is 225 x 225 μm in size, scale bar = 20 μm.
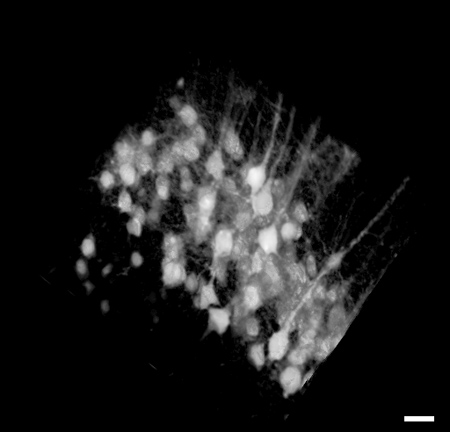
Figure 4. Representative image of 3D reconstruction (225 x 225 x 320 μm) of the image stack in Figure 3. Image is 225 x 225 μm in size, scale bar = 28 μm.
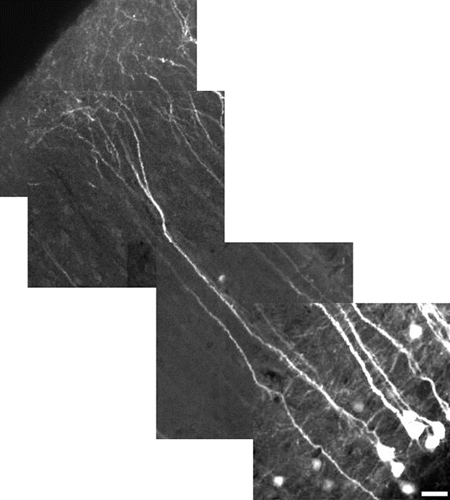
Figure 5. High-resolution reconstructed image of a layer V pyramidal neuron of the neocortex using a 10X digital zoom. Neuron measures 485 μm in length, scale bar = 25 μm. The field of view for each image tile is 180 μm x 180 μm. Each tile is at a different depth (z-level). The cortical apical dendrites do not, in general, express YFP as well as the soma. To have both in the same field of view, as shown here, the power was lowered to minimize saturation of soma; this makes the fine process look dimmer. Our emphasis here was on demonstrating field-of-view rather than fine detail. To reveal finer details, use a digital zoom, focus on a region without soma, and increase the laser power.
