RNA-seq wurde im Laufe der Jahre häufig verwendet, typischerweise zur Schätzung der differentiellen Genexpression und Genentdeckung1. Darüber hinaus kann es auch verwendet werden, um die unterschiedliche Nutzung auf Exon-Ebene aufgrund der Genexprimierung verschiedener Isoformen abzuschätzen, was zu einem besseren Verständnis der Genregulation auf posttranskriptioneller Ebene beiträgt. Die Mehrheit der eukaryotischen Gene erzeugt verschiedene Isoformen durch alternatives Spleißen (AS), um die Vielfalt der mRNA-Expression zu erhöhen. AS-Ereignisse können in verschiedene Muster unterteilt werden: Überspringen vollständiger Exons (SE), bei denen ein (“Kassetten”) Exon zusammen mit seinen flankierenden Introns vollständig aus dem Transkript entfernt wird; alternative (Donor) 5′-Spleißstellenauswahl (A5SS) und alternative 3′ (Akzeptor) Spleißstellenauswahl (A3SS), wenn zwei oder mehr Spleißstellen an beiden Enden eines Exons vorhanden sind; Beibehaltung von Introns (RI), wenn ein Intron innerhalb des reifen mRNA-Transkripts beibehalten wird, und gegenseitiger Ausschluss der Exon-Nutzung (MXE), wobei nur eines der beiden verfügbaren Exons gleichzeitig beibehalten werden kann 2,3. Die alternative Polyadenylierung (APA) spielt auch eine wichtige Rolle bei der Regulierung der Genexpression unter Verwendung alternativer Poly(A)-Stellen, um mehrere mRNA-Isoformen aus einem einzigen Transkriptzu erzeugen 4. Die meisten Polyadenylierungsstellen (pAs) befinden sich in der 3′ untranslatierten Region (3′ UTRs) und erzeugen mRNA-Isoformen mit unterschiedlichen 3′ UTR-Längen. Da die 3′ UTR die zentrale Drehscheibe für die Erkennung regulatorischer Elemente ist, können unterschiedliche 3′ UTR-Längen die mRNA-Lokalisierung, Stabilität und Translation beeinflussen5. Es gibt eine Klasse von 3′-Endsequenzierungsassays, die für den Nachweis von APA optimiert sind und sich in den Details des Protokolls6 unterscheiden. Die hier beschriebene Pipeline ist für PolyA-seq ausgelegt, kann aber wie beschrieben für andere Protokolle angepasst werden.
In dieser Studie stellen wir eine Pipeline von differentiellen Exon-Analysemethoden7,8 (Abbildung 1) vor, die in zwei große Kategorien unterteilt werden können: exon-basiert (DEXSeq9, diffSplice 10) und ereignisbasiert (replicate Multivariate Analysis of Transcript Splicing (rMATS)11). Die Exon-basierten Methoden vergleichen die Faltenänderung über die Bedingungen einzelner Exons hinweg mit einem Maß für die gesamte Genfaltenänderung, um differentiell exprimierte Exon-Nutzung zu nennen, und berechnen daraus ein Maß für die AS-Aktivität auf Genebene. Ereignisbasierte Methoden verwenden Exon-Intron-Spanning-Junction-Lesevorgänge, um bestimmte Spleißereignisse wie Exon-Skipping oder Beibehaltung von Introns zu erkennen und zu klassifizieren und diese AS-Typen in Ausgabe3 zu unterscheiden. Somit bieten diese Methoden komplementäre Sichtweisen für eine vollständige Analyse von AS12,13. Wir haben DEXSeq (basierend auf dem DESeq214 DGE-Paket) und diffSplice (basierend auf dem Limma10 DGE-Paket) für die Studie ausgewählt, da sie zu den am häufigsten verwendeten Paketen für die differentielle Spleißanalyse gehören. rMATS wurde als beliebte Methode für die ereignisbasierte Analyse ausgewählt. Eine weitere beliebte ereignisbasierte Methode ist MISO (Mixture of Isoforms)1. Für APA adaptieren wir den Exon-basierten Ansatz.
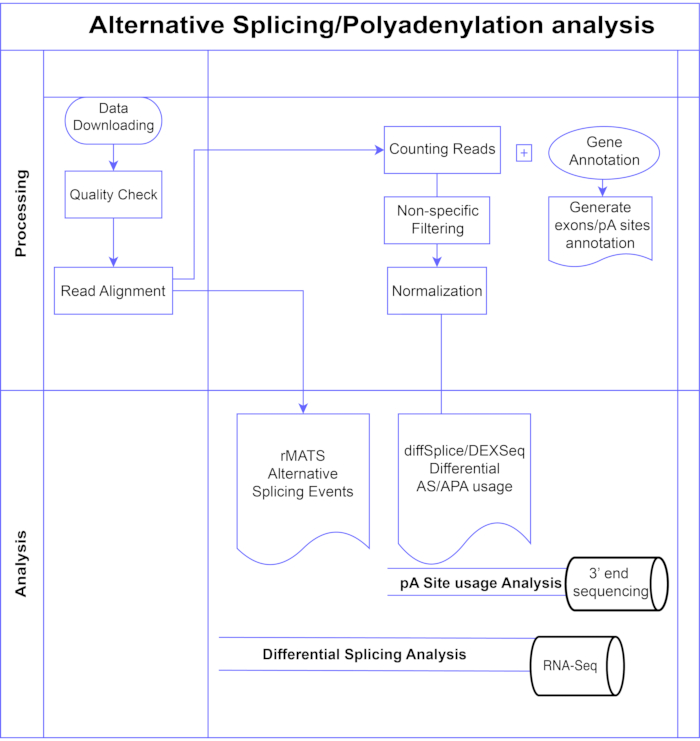
Abbildung 1. Analyse-Pipeline. Flussdiagramm der in der Analyse verwendeten Schritte. Zu den Schritten gehören: Abrufen der Daten, Durchführen von Qualitätsprüfungen und Leseausrichtung, gefolgt von Zählen von Lesevorgängen unter Verwendung von Anmerkungen für bekannte Exons, Introns und pA-Stellen, Filtern zur Entfernung niedriger Zählungen und Normalisierung. PolyA-seq-Daten wurden für alternative pA-Stellen mit diffSplice/DEXSeq-Methoden analysiert, Bulk-RNA-Seq wurde auf alternative Spleißung auf Exon-Ebene mit diffSplice/DEXseq-Methoden analysiert und AS-Ereignisse mit rMATS analysiert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
Die in dieser Umfrage verwendeten RNA-seq-Daten stammen von Gene Expression Omnibus (GEO) (GSE138691)15. Wir verwendeten Maus-RNA-seq-Daten aus dieser Studie mit zwei Bedingungsgruppen: Wildtyp (WT) und Muskelblind-ähnlicher Typ-1-Knockout (Mbnl1 KO) mit jeweils drei Replikaten. Um die Analyse der differentiellen Polyadenylierungsstandortnutzung zu demonstrieren, erhielten wir PolyA-seq-Daten von Mausembryo-Fibroblasten (MEFs) (GEO Accession GSE60487)16. Die Daten haben vier Bedingungsgruppen: Wild-Typ (WT), Muskelblind-Typ 1/Typ 2 Doppel-Knockout (Mbnl1/2 DKO), Mbnl 1/2 DKO mit Mbnl3-Knockdown (KD) und Mbnl1/2 DKO mit Mbnl3-Kontrolle (Strg). Jede Bedingungsgruppe besteht aus zwei Replikaten.
| GEO-Beitritt | SRA-Ausführungsnummer | Name des Beispiels | Zustand | Replizieren | Gewebe | Sequenzierung | Leselänge | |
| RNA-Seq | GSM4116218 | SRR10261601 | Mbnl1KO_Thymus_1 | Mbnl1 Knockout | Wiederholung 1 | Thymus | Gepaartes Ende | 100 bp |
| GSM4116219 | SRR10261602 | Mbnl1KO_Thymus_2 | Mbnl1 Knockout | Wiederholung 2 | Thymus | Gepaartes Ende | 100 bp | |
| GSM4116220 | SRR10261603 | Mbnl1KO_Thymus_3 | Mbnl1 Knockout | Wiederholung 3 | Thymus | Gepaartes Ende | 100 bp | |
| GSM4116221 | SRR10261604 | WT_Thymus_1 | Wildtyp | Wiederholung 1 | Thymus | Gepaartes Ende | 100 bp | |
| GSM4116222 | SRR10261605 | WT_Thymus_2 | Wildtyp | Wiederholung 2 | Thymus | Gepaartes Ende | 100 bp | |
| GSM4116223 | SRR10261606 | WT_Thymus_3 | Wildtyp | Wiederholung 3 | Thymus | Gepaartes Ende | 100 bp | |
| 3P-Seq | GSM1480973 | SRR1553129 | WT_1 | Wildtyp (WT) | Wiederholung 1 | Embryonale Fibroblasten (MEFs) der Maus | Single-End | 40 bp |
| GSM1480974 | SRR1553130 | WT_2 | Wildtyp (WT) | Wiederholung 2 | Embryonale Fibroblasten (MEFs) der Maus | Single-End | 40 bp | |
| GSM1480975 | SRR1553131 | DKO_1 | Mbnl 1/2 Doppel-Knockout (DKO) | Wiederholung 1 | Embryonale Fibroblasten (MEFs) der Maus | Single-End | 40 bp | |
| GSM1480976 | SRR1553132 | DKO_2 | Mbnl 1/2 Doppel-Knockout (DKO) | Wiederholung 2 | Embryonale Fibroblasten (MEFs) der Maus | Single-End | 40 bp | |
| GSM1480977 | SRR1553133 | DKOsiRNA_1 | Mbnl 1/2 Doppel-Knockout mit Mbnl 3 siRNA (KD) | Wiederholung 1 | Embryonale Fibroblasten (MEFs) der Maus | Single-End | 40 bp | |
| GSM1480978 | SRR1553134 | DKOsiRNA_2 | Mbnl 1/2 Doppel-Knockout mit Mbnl 3 siRNA (KD) | Wiederholung 2 | Embryonale Fibroblasten (MEFs) der Maus | Single-End | 36 bp | |
| GSM1480979 | SRR1553135 | DKONTsiRNA_1 | Mbnl 1/2 Doppel-Knockout mit nicht-targetender siRNA (Ctrl) | Wiederholung 1 | Embryonale Fibroblasten (MEFs) der Maus | Single-End | 40 bp | |
| GSM1480980 | SRR1553136 | DKONTsiRNA_2 | Mbnl 1/2 Doppel-Knockout mit nicht-targetender siRNA (Ctrl) | Wiederholung 2 | Embryonale Fibroblasten (MEFs) der Maus | Single-End | 40 bp |
Tabelle 1. Zusammenfassung der RNA-Seq- und PolyA-seq-Datensätze, die für die Analyse verwendet wurden.
