RNA-seq היה בשימוש נרחב לאורך השנים בדרך כלל להערכת ביטוי גנים דיפרנציאליים וגילוי גנים1. בנוסף, ניתן להשתמש בו גם כדי להעריך שימוש משתנה ברמת אקסון עקב גנים המבטאים איזופורמים שונים, ובכך לתרום להבנה טובה יותר של ויסות גנים ברמה שלאחר השעתוק. רוב הגנים האאוקריוטים מייצרים איזופורמים שונים על ידי שחבור חלופי (AS) כדי להגדיל את המגוון של ביטוי mRNA. ניתן לחלק את אירועי AS לדפוסים שונים: דילוג על אקסונים שלמים (SE) שבהם אקסון (“קלטת”) מוסר לחלוטין מהתמליל יחד עם האינטרונים האגפים שלו; בחירת אתר שחבור חלופי (תורם) 5′ (A5SS) וחלופה 3′ (מקבל) בחירת אתר שחבור (A3SS) כאשר שני אתרי שחבור או יותר נמצאים משני קצותיו של אקסון; שמירה של אינטרונים (RI) כאשר אינטרון נשמר בתוך תעתיק mRNA בוגר והדרה הדדית של שימוש באקסון (MXE) כאשר רק אחד משני האקסונים הזמינים יכול להישמר בכל פעם 2,3. פוליאדנילציה חלופית (APA) ממלאת גם תפקיד חשוב בוויסות ביטוי גנים באמצעות אתרי פולי (A) חלופיים ליצירת איזופורמים מרובים של mRNA מתעתיק יחיד4. רוב אתרי הפוליאדנילציה (pAs) ממוקמים באזור 3′ לא מתורגם (3′ UTRs), ומייצרים איזופורמים של mRNA עם אורכי UTR מגוונים של 3′. מכיוון שה-UTR 3′ הוא הרכזת המרכזית לזיהוי אלמנטים רגולטוריים, אורכי UTR שונים של 3′ יכולים להשפיע על לוקליזציה, יציבות ותרגוםmRNA 5. ישנם סוגים של מבחני רצף קצה 3 ‘הממוטבים לזיהוי APA השונים בפרטי הפרוטוקול6. הצינור המתואר כאן מיועד ל- PolyA-seq, אך ניתן להתאים אותו לפרוטוקולים אחרים כמתואר.
במחקר זה אנו מציגים צינור של שיטות ניתוח אקסון דיפרנציאליות7,8 (איור 1), שניתן לחלק לשתי קטגוריות רחבות: מבוססות אקסון (DEXSeq9, diffSplice 10) ומבוססות אירועים (ניתוח רב-משתני משוכפל של שחבור תעתיק (rMATS)11). השיטות המבוססות על אקסון משוות את שינוי הקיפול על פני תנאים של אקסונים בודדים, כנגד מידה של שינוי קיפול גנים כולל כדי לקרוא לשימוש באקסון המתבטא באופן דיפרנציאלי, ומתוך כך מחשבים מדידה ברמת הגן של פעילות AS. שיטות מבוססות אירועים משתמשות בקריאות צומת exon-intron-spanning כדי לזהות ולסווג אירועי שחבור ספציפיים כגון דילוג אקסון או שמירה של אינטרונים, ולהבחין בין סוגי AS אלה בפלט3. לפיכך, שיטות אלה מספקות תצוגות משלימות לניתוח מלא של AS12,13. בחרנו ב- DEXSeq (בהתבסס על חבילת DESeq214 DGE) ו- diffSplice (בהתבסס על חבילת Limma10 DGE) למחקר מכיוון שהם בין החבילות הנפוצות ביותר לניתוח שחבור דיפרנציאלי. rMATS נבחרה כשיטה פופולרית לניתוח מבוסס אירועים. שיטה פופולרית נוספת המבוססת על אירועים היא MISO (תערובת של איזופורמים)1. עבור APA אנו מתאימים את הגישה מבוססת exon.
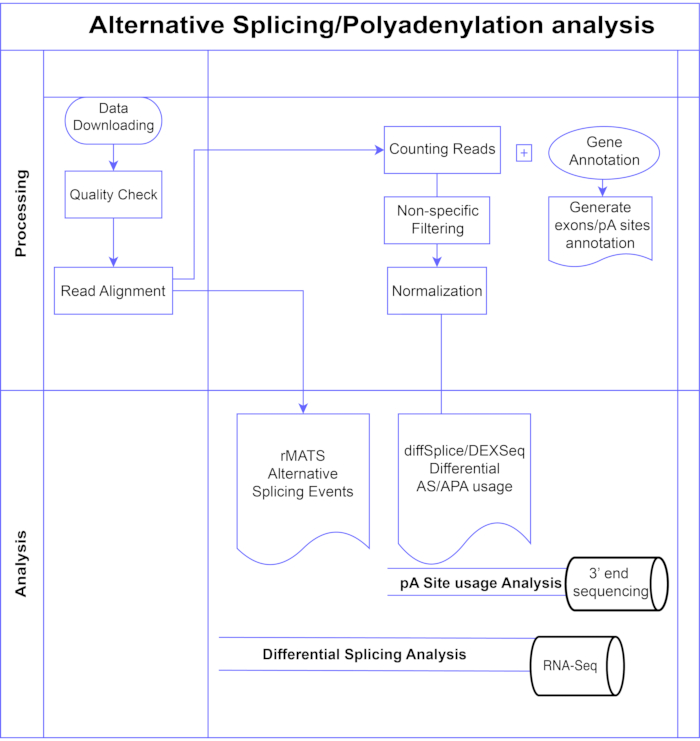
איור 1. צינור ניתוח. תרשים זרימה של השלבים המשמשים בניתוח. השלבים כוללים: קבלת הנתונים, ביצוע בדיקות איכות ויישור קריאה ולאחר מכן ספירת קריאות באמצעות ביאורים עבור exons, introns ואתרי pA ידועים, סינון כדי להסיר ספירות נמוכות ונורמליזציה. נתוני PolyA-seq נותחו עבור אתרי pA חלופיים באמצעות שיטות diffSplice/DEXSeq, RNA-Seq בתפזורת נותח עבור שחבור חלופי ברמת האקסון בשיטות diffSplice/DEXseq, ואירועי AS נותחו עם rMATS. אנא לחץ כאן כדי להציג גרסה גדולה יותר של נתון זה.
נתוני ה-RNA-seq ששימשו בסקר זה נרכשו מביטוי גנים אומניבוס (GEO) (GSE138691)15. השתמשנו בנתוני RNA-seq של עכברים ממחקר זה עם שתי קבוצות מצב: נוקאאוט מסוג פראי (WT) ונוקאאוט דמוי שריר מסוג 1 (Mbnl1 KO) עם שלושה שכפולים כל אחד. כדי להדגים ניתוח שימוש באתר פוליאדנילציה דיפרנציאלית, השגנו נתוני פוליA-seq של עוברי עכברים (MEFs) (GEO Accession GSE60487)16. לנתונים יש ארבע קבוצות מצבים: סוג פראי (WT), דמוי שרירים מסוג 1/סוג 2 נוקאאוט כפול (Mbnl1/2 DKO), Mbnl 1/2 DKO עם נוקאאוט Mbnl3 (KD) ו- Mbnl1/2 DKO עם בקרת Mbnl3 (Ctrl). כל קבוצת תנאים מורכבת משני עותקים משוכפלים.
| הצטרפות גיאוגרפית | מספר הפעלה של SRA | שם לדוגמה | תנאי | לשכפל | טישו | רצף | אורך קריאה | |
| רנ”א-סק | GSM4116218 | SRR10261601 | Mbnl1KO_Thymus_1 | נוקאאוט Mbnl1 | נציג 1 | התימוס | סוף מזווג | 100 כ”ס |
| GSM4116219 | SRR10261602 | Mbnl1KO_Thymus_2 | נוקאאוט Mbnl1 | חזרה 2 | התימוס | סוף מזווג | 100 כ”ס | |
| GSM4116220 | SRR10261603 | Mbnl1KO_Thymus_3 | נוקאאוט Mbnl1 | חזרה 3 | התימוס | סוף מזווג | 100 כ”ס | |
| GSM4116221 | SRR10261604 | WT_Thymus_1 | סוג פראי | נציג 1 | התימוס | סוף מזווג | 100 כ”ס | |
| GSM4116222 | SRR10261605 | WT_Thymus_2 | סוג פראי | חזרה 2 | התימוס | סוף מזווג | 100 כ”ס | |
| GSM4116223 | SRR10261606 | WT_Thymus_3 | סוג פראי | חזרה 3 | התימוס | סוף מזווג | 100 כ”ס | |
| 3P-Seq | GSM1480973 | SRR1553129 | WT_1 | סוג פראי (WT) | נציג 1 | פיברובלסטים עובריים לעכבר (MEFs) | קצה יחיד | 40 bp |
| GSM1480974 | SRR1553130 | WT_2 | סוג פראי (WT) | חזרה 2 | פיברובלסטים עובריים לעכבר (MEFs) | קצה יחיד | 40 bp | |
| GSM1480975 | SRR1553131 | DKO_1 | Mbnl 1/2 נוקאאוט כפול (DKO) | נציג 1 | פיברובלסטים עובריים לעכבר (MEFs) | קצה יחיד | 40 bp | |
| GSM1480976 | SRR1553132 | DKO_2 | Mbnl 1/2 נוקאאוט כפול (DKO) | חזרה 2 | פיברובלסטים עובריים לעכבר (MEFs) | קצה יחיד | 40 bp | |
| GSM1480977 | SRR1553133 | DKOsiRNA_1 | Mbnl 1/2 נוקאאוט כפול עם Mbnl 3 siRNA (KD) | נציג 1 | פיברובלסטים עובריים לעכבר (MEFs) | קצה יחיד | 40 bp | |
| GSM1480978 | SRR1553134 | DKOsiRNA_2 | Mbnl 1/2 נוקאאוט כפול עם Mbnl 3 siRNA (KD) | חזרה 2 | פיברובלסטים עובריים לעכבר (MEFs) | קצה יחיד | 36 bp | |
| GSM1480979 | SRR1553135 | DKONTsiRNA_1 | Mbnl 1/2 נוקאאוט כפול עם siRNA ללא מיקוד (Ctrl) | נציג 1 | פיברובלסטים עובריים לעכבר (MEFs) | קצה יחיד | 40 bp | |
| GSM1480980 | SRR1553136 | DKONTsiRNA_2 | Mbnl 1/2 נוקאאוט כפול עם siRNA ללא מיקוד (Ctrl) | חזרה 2 | פיברובלסטים עובריים לעכבר (MEFs) | קצה יחיד | 40 bp |
טבלה 1. סיכום של מערכי נתונים של RNA-Seq ו- PolyA-seq המשמשים לניתוח.
