O RNA-seq tem sido amplamente utilizado ao longo dos anos, tipicamente para estimar a expressão gênica diferencial e a descoberta de genes1. Além disso, também pode ser utilizado para estimar o uso variável do nível de éxons devido ao gene que expressa diferentes isoformas, contribuindo assim para uma melhor compreensão da regulação gênica no nível pós-transcricional. A maioria dos genes eucarióticos gera diferentes isoformas por splicing alternativo (AS) para aumentar a diversidade de expressão de mRNA. Os eventos AS podem ser divididos em diferentes padrões: pulando éxons completos (SE), onde um éxon (“”) é completamente removido da transcrição junto com seus íntrons flanqueadores; seleção alternativa (doador) de local de emenda de 5′ (A5SS) e seleção de local de emenda alternativa de 3′ (aceitador) (A3SS) quando dois ou mais locais de emenda estiverem presentes em cada extremidade de um éxon; retenção de íntrons (IR) quando um íntron é retido dentro do transcrito de mRNA maduro e exclusão mútua do uso de éxons (MXE) onde apenas um dos dois éxons disponíveis pode ser retido de cada vez 2,3. A poliadenilação alternativa (APA) também desempenha um papel importante na regulação da expressão gênica usando sítios alternativos de poli (A) para gerar múltiplas isoformas de mRNA a partir de um único transcrito4. A maioria dos sítios de poliadenilação (pAs) está localizada na região não traduzida de 3′ (UTRs de 3′), gerando isoformas de mRNA com diversos comprimentos UTR de 3′. Como a UTR de 3′ é o hub central para o reconhecimento de elementos regulatórios, diferentes comprimentos de UTR de 3′ podem afetar a localização, a estabilidade e a tradução do mRNA5. Há uma classe de ensaios de sequenciamento final de 3′ otimizados para detectar APA que diferem nos detalhes do protocolo6. O pipeline descrito aqui é projetado para PolyA-seq, mas pode ser adaptado para outros protocolos, conforme descrito.
Neste estudo, apresentamos um pipeline de métodos diferenciais de análise de éxons7,8 (Figura 1), que podem ser divididos em duas grandes categorias: baseada em éxons (DEXSeq9, diffSplice 10) e baseada em eventos (replicate Multivariate Analysis of Transcript Splicing (rMATS)11). Os métodos baseados em éxons comparam a mudança de dobra entre as condições de éxons individuais, contra uma medida de mudança geral de dobra gênica para chamar o uso de éxons diferencialmente expressos e, a partir disso, calculam uma medida em nível de gene da atividade EA. Os métodos baseados em eventos usam leituras de junção de abrangência de exon-intron para detectar e classificar eventos de splicing específicos, como pulo de éxon ou retenção de íntrons, e distinguir esses tipos de AS na saída3. Assim, esses métodos fornecem visões complementares para uma análise completa da EA12,13. Selecionamos o DEXSeq (baseado no pacote DESeq214 DGE) e o diffSplice (baseado no pacote Limma10 DGE) para o estudo, pois estão entre os pacotes mais utilizados para análise de splicing diferencial. O rMATS foi escolhido como um método popular para análise baseada em eventos. Outro método popular baseado em eventos é o MISO (Mix of Isoforms)1. Para a APA, adaptamos a abordagem baseada em exons.
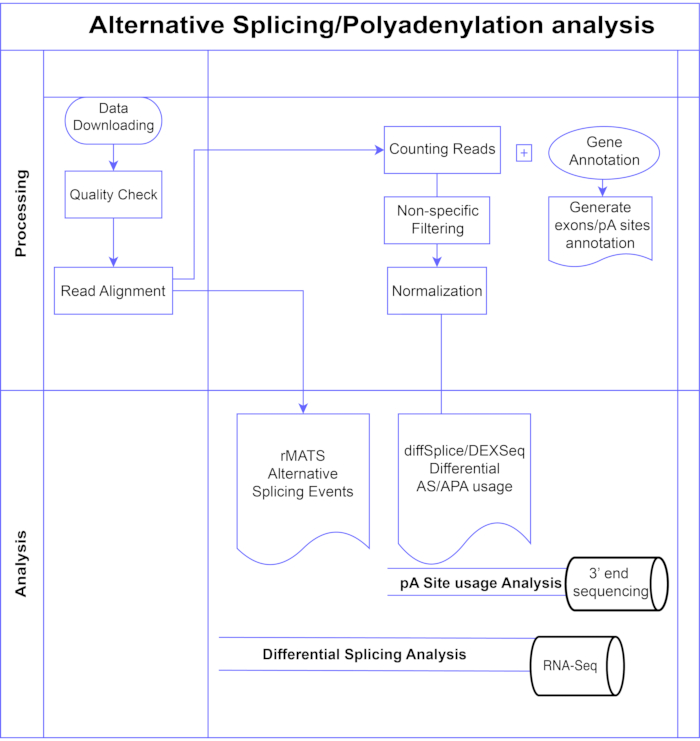
Figura 1. Análise de pipeline. Fluxograma das etapas utilizadas na análise. As etapas incluem: obtenção dos dados, realização de verificações de qualidade e alinhamento de leitura seguidas de contagem de leituras usando anotações para éxons, íntrons e sites pA conhecidos, filtragem para remover contagens baixas e normalização. Os dados de PolyA-seq foram analisados para sítios alternativos de pA usando os métodos diffSplice/DEXSeq, o RNA-Seq em massa foi analisado para splicing alternativo no nível do éxon com os métodos diffSplice/DEXseq e os eventos AS analisados com rMATS. Por favor, clique aqui para ver uma versão maior desta figura.
Os dados de RNA-seq utilizados neste levantamento foram adquiridos do Gene Expression Omnibus (GEO) (GSE138691)15. Utilizamos dados de RNA-seq de camundongos deste estudo com dois grupos de condições: wild-type (WT) e Muscleblind-like type 1 knockout (Mbnl1 KO) com três repetições cada. Para demonstrar a análise diferencial do uso do sítio de poliadenilação, obtivemos dados PolyA-seq de fibroblastos embrionários de camundongos (MEFs) (GEO Accession GSE60487)16. Os dados têm quatro grupos de condições: Wild-type (WT), Muscleblind-like type1/type 2 double knockout (Mbnl1/2 DKO), Mbnl 1/2 DKO com Mbnl3 knockdown (KD) e Mbnl1/2 DKO com controle Mbnl3 (Ctrl). Cada grupo de condições consiste em duas replicações.
| Adesão ao GEO | Número de execução SRA | Nome do exemplo | Condição | Replicar | Tecido | Seqüenciamento | Comprimento de leitura | |
| RNA-Seq | Telemóvel GSM4116218 | SRR10261601 | Mbnl1KO_Thymus_1 | Nocaute Mbnl1 | Rep 1 | Timo | Extremidade emparelhada | 100 pb |
| Telemóvel GSM4116219 | SRR10261602 | Mbnl1KO_Thymus_2 | Nocaute Mbnl1 | Rep 2 | Timo | Extremidade emparelhada | 100 pb | |
| Telemóvel GSM4116220 | SRR10261603 | Mbnl1KO_Thymus_3 | Nocaute Mbnl1 | Rep 3 | Timo | Extremidade emparelhada | 100 pb | |
| Telemóvel GSM4116221 | SRR10261604 | WT_Thymus_1 | Tipo selvagem | Rep 1 | Timo | Extremidade emparelhada | 100 pb | |
| Telemóvel GSM4116222 | SRR10261605 | WT_Thymus_2 | Tipo selvagem | Rep 2 | Timo | Extremidade emparelhada | 100 pb | |
| Telemóvel GSM4116223 | SRR10261606 | WT_Thymus_3 | Tipo selvagem | Rep 3 | Timo | Extremidade emparelhada | 100 pb | |
| 3P-Seq | Mensagem GSM1480973 | SRR1553129 | WT_1 | Tipo selvagem (WT) | Rep 1 | Fibroblastos embrionários de camundongos (MEFs) | Extremidade única | 40 pb |
| Telemóvel 1480974 | SRR1553130 | WT_2 | Tipo selvagem (WT) | Rep 2 | Fibroblastos embrionários de camundongos (MEFs) | Extremidade única | 40 pb | |
| Telemóvel 1480975 | SRR1553131 | DKO_1 | Mbnl 1/2 nocaute duplo (DKO) | Rep 1 | Fibroblastos embrionários de camundongos (MEFs) | Extremidade única | 40 pb | |
| Mensagem GSM1480976 | SRR1553132 | DKO_2 | Mbnl 1/2 nocaute duplo (DKO) | Rep 2 | Fibroblastos embrionários de camundongos (MEFs) | Extremidade única | 40 pb | |
| Telemóvel 1480977 | SRR1553133 | DKOsiRNA_1 | Mbnl 1/2 nocaute duplo com Mbnl 3 siRNA (KD) | Rep 1 | Fibroblastos embrionários de camundongos (MEFs) | Extremidade única | 40 pb | |
| Telemóvel 1480978 | SRR1553134 | DKOsiRNA_2 | Mbnl 1/2 nocaute duplo com Mbnl 3 siRNA (KD) | Rep 2 | Fibroblastos embrionários de camundongos (MEFs) | Extremidade única | 36 pb | |
| Telemóvel 1480979 | SRR1553135 | DKONTsiRNA_1 | Mbnl 1/2 nocaute duplo com siRNA não direcionado (Ctrl) | Rep 1 | Fibroblastos embrionários de camundongos (MEFs) | Extremidade única | 40 pb | |
| Telemóvel 1480980 | SRR1553136 | DKONTsiRNA_2 | Mbnl 1/2 nocaute duplo com siRNA não direcionado (Ctrl) | Rep 2 | Fibroblastos embrionários de camundongos (MEFs) | Extremidade única | 40 pb |
Tabela 1. Resumo dos conjuntos de dados de RNA-Seq e PolyA-seq utilizados para a análise.
