Au cours des deux dernières décennies, les produits biothérapeutiques ont évolué pour devenir un pilier de l’industrie pharmaceutique moderne. La pandémie de SRAS-CoV2 et d’autres maladies potentiellement mortelles ont encore accru la nécessité d’un développement plus rapide et plus large de molécules biopharmaceutiques 1,2,3.
Le poids moléculaire biothérapeutique est critique pour l’identification de la molécule, en combinaison avec d’autres tests analytiques. Les masses de sous-unités intactes et réduites sont utilisées tout au long des cycles de vie de découverte et de développement dans le cadre de stratégies de contrôle visant à maintenir la qualité, comme décrit dans le QTPP (Quality Target Product Profile)4.
Le développement analytique dans l’industrie biopharmaceutique repose fortement sur les mesures de masse pour l’analyse de la masse intacte et la caractérisation en profondeur à l’aide de la cartographie peptidique ou de la surveillance par méthode multi-attributs (MAM). Au centre de ces techniques utilisant des plates-formes modernes de spectrométrie de masse (MS) se trouve la capacité de fournir des mesures de masse précises à haute résolution (HR/AM). La plupart des instruments HR/AM offrent des précisions de masse comprises entre 0,5 et 5 ppm, qui s’adaptent à la plage de masse. La capacité de mesurer avec précision les masses de grosses molécules intactes permet d’identifier rapidement et en toute confiance les produits thérapeutiques à grandes molécules. Comme la résolution isotopique ne peut pas être atteinte dans des conditions expérimentales typiques pour les grosses molécules (>10 kDa), les masses moyennes doivent être calculées pour la comparaison et l’identification 5,6.
Un spectre de masse protéique intact ou sous-unitaire typique représente le profil protéoforme global, qui contient des informations composites sur les différentes formes moléculaires résultant des modifications post-traductionnelles (PTM) et de toute différence de structure primaire, telle que les clips ou les variantes de séquence. La facilité et le débit relativement élevés de ces mesures les rendent attrayantes pour la caractérisation et les contrôles de surveillance en cours de processus 7,8. L’analyse des données pour ces expériences nécessite généralement que l’utilisateur définisse l’espace de recherche des formes moléculaires (gamme de PTM ou d’autres formes moléculaires). Pour les protéines glycosylées, cet espace de recherche est largement déterminé par l’hétérogénéité des glycoformes. Les combinaisons de plusieurs PTM, de configurations de liaisons disulfure et d’autres variations le long de la structure primaire rendent le calcul de toutes les formes moléculaires possibles fastidieux. Par conséquent, le calcul manuel des formes moléculaires possibles est un processus qui prend du temps et des ressources et qui présente un fort potentiel d’erreur humaine.
Nous présentons ici un outil de calcul de masse qui a été développé en tenant compte des caractéristiques les plus importantes des molécules biothérapeutiques, telles que les anticorps monoclonaux, les bsAb, les ADC, etc. L’outil permet d’incorporer facilement des variables de l’espace de recherche pour le calcul cohérent des masses et des compositions élémentaires. La nature modulaire de cet outil permettra de le développer et de l’appliquer au calcul de masse et à l’appariement de masse pour d’autres modalités.
Le module d’interface graphique permet à l’utilisateur de spécifier l’entrée pour le calcul de la masse, comme illustré à la figure 1 ; Plus précisément, l’utilisateur saisit des séquences d’acides aminés d’une seule lettre pour les chaînes d’anticorps légères et lourdes. Les modifications courantes pour la cyclisation N-terminale à chaîne lourde et l’écrêtage de la lysine C-terminale sont incluses sous forme de cases à cocher. De plus, la formule chimique/la composition élémentaire peut être ajoutée/soustraite de ces chaînes protéiques via la zone de texte Chem Mod correspondante. Cela permet à l’utilisateur d’ajouter une composition élémentaire qui comprend de multiples modifications post-traductionnelles ou une charge utile de petites molécules dans le cas d’un ADC. Comme la plupart des anticorps monoclonaux thérapeutiques sont conçus pour éliminer les sites de glycosylation dans la chaîne légère, la glycosylation dans la chaîne légère est facultative et peut être spécifiée à l’aide d’une case à cocher sur l’interface graphique.
Une variante typique de l’analyse de la masse intacte pour les anticorps est une analyse de masse sous-unitaire réduite, où la chaîne légère est détachée de la chaîne lourde en réduisant les liaisons disulfure interchaînes. Selon la force de l’agent réducteur utilisé, les liaisons disulfure intra-chaîne peuvent être clivées ou non. Les utilisateurs ont la possibilité d’entrer le nombre total de liaisons disulfure en fonction du sous-type d’IgG ou dans le cas d’un ADC9 conjugué à la cystéine.
L’application calcule les masses de manière ascendante, dans laquelle les compositions élémentaires sont d’abord calculées pour les chaînes lourdes et les chaînes légères individuelles. Ensuite, l’écrêtage de Lys de la cyclisation N-terminale à chaîne lourde (HC) est pris en compte en ajustant les compositions élémentaires calculées. Toutes les modifications chimiques spécifiées sont ensuite appliquées aux chaînes lourdes et/ou légères. En fonction du type d’analyse et des modèles de liaison disulfure spécifiés par l’utilisateur, le nombre d’hydrogènes est ajusté pour les deux chaînes polypeptidiques. Les masses glycosylées HC et de chaîne légère (LC) (en option) sont calculées en fonction de l’entrée de l’utilisateur. Enfin, plusieurs masses HC et LC sont combinées, et les numéros de liaison disulfure sont automatiquement mis à jour pour le calcul de la masse intacte.
Avec des molécules plus grosses telles que des protéines intactes, les masses monoisotopiques ne peuvent pas être mesurées en raison du défaut de masse additif lors de l’utilisation de spectromètres de masse avec un pouvoir de résolution typique. Au lieu de cela, les masses nominales ou moyennes sont mesurées ou déclarées 5,10,11,12,13. Les masses élémentaires moyennes peuvent varier en fonction de la source utilisée pour les masses sélectionnées14,15. Bien que les différences dans les masses élémentaires puissent être faibles, elles peuvent s’additionner pour atteindre des valeurs significatives pour les calculs de poids moléculaire de grosses molécules. Les masses élémentaires moyennes utilisées par défaut dans l’application logicielle sont indiquées dans le tableau supplémentaire 1. Pour les environnements réglementés comme le domaine de la recherche et du développement (R-D) biopharmaceutique, il est important de maintenir des masses moléculaires constantes, car les changements de masse peuvent impliquer des changements dans l’entité moléculaire lors des dépôts réglementaires. Pour assurer la cohérence de l’utilisation des masses élémentaires, un dictionnaire des masses élémentaires est inclus avec l’outil logiciel sous la forme d’un fichier texte csv (valeurs séparées par des virgules) : Element_Mass.csv (Fichier de codage supplémentaire 1). De même, une liste organisée des compositions de glycanes généralement observées sur les anticorps monoclonaux est incluse : Glycan.csv (Supplementary Coding File 2). Les deux fichiers sont enregistrés dans le même dossier qu’une application exécutable et peuvent être modifiés par l’utilisateur pour utiliser une liste de masse élémentaire spécifique ou une bibliothèque de glycanes.
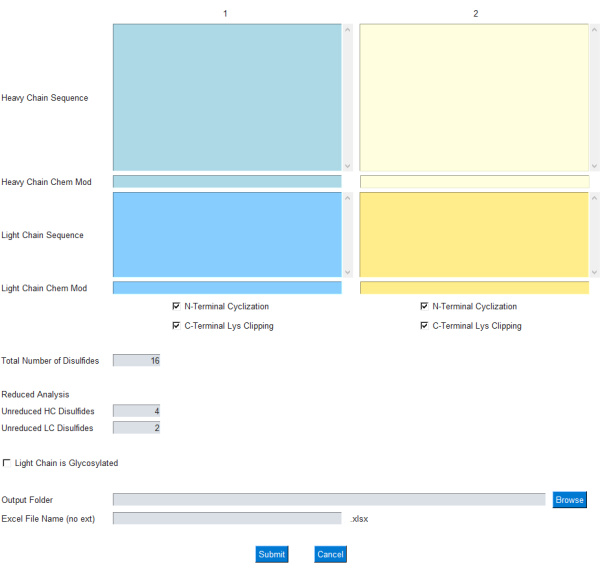
Figure 1 : Interface graphique de l’application mAbScale. Le module GUI permet à l’utilisateur de spécifier l’entrée pour le calcul de la masse. L’utilisateur saisit des séquences d’acides aminés d’une seule lettre pour les chaînes d’anticorps légères et lourdes. Les modifications courantes pour la cyclisation N-terminale à chaîne lourde et l’écrêtage de la lysine C-terminale sont incluses sous forme de cases à cocher. Les formules chimiques/compositions élémentaires peuvent être ajoutées/soustraites via la zone de texte Chem Mod correspondante. Veuillez cliquer ici pour voir une version agrandie de cette figure.
