Negli ultimi due decenni, la bioterapia si è evoluta fino a diventare un pilastro della moderna industria farmaceutica. La pandemia di SARS-CoV2 e altre condizioni potenzialmente letali hanno ulteriormente aumentato la necessità di uno sviluppo più rapido e più ampio di molecole biofarmaceutiche 1,2,3.
Il peso molecolare bioterapeutico è fondamentale per l’identificazione della molecola, in combinazione con altri saggi analitici. Le masse delle subunità intatte e ridotte sono utilizzate durante tutto il ciclo di vita della scoperta e dello sviluppo come parte delle strategie di controllo volte a mantenere la qualità, come descritto nel QTPP (Quality Target Product Profile)4.
Lo sviluppo analitico nell’industria biofarmaceutica si basa in larga misura sulle misurazioni di massa per l’analisi della massa intatta e la caratterizzazione profonda utilizzando la mappatura dei peptidi o il monitoraggio con il metodo multi-attributo (MAM). Al centro di queste tecniche che utilizzano le moderne piattaforme di spettrometria di massa (MS) c’è la capacità di fornire misurazioni di massa accurate ad alta risoluzione (HR/AM). La maggior parte degli strumenti HR/AM produce precisioni di massa nell’intervallo di 0,5-5 ppm, che scalano con l’intervallo di massa. La capacità di misurare con precisione le masse per le grandi molecole intatte consente l’identificazione rapida e sicura delle terapie a grandi molecole. Poiché la risoluzione isotopica non può essere raggiunta utilizzando le condizioni sperimentali tipiche per molecole di grandi dimensioni (>10 kDa), è necessario calcolare le masse medie per il confronto e l’identificazione 5,6.
Un tipico spettro di massa di proteine intatte o subunità rappresenta il profilo complessivo della proteoforma, che contiene informazioni composite sulle varie forme molecolari risultanti da modificazioni post-traduzionali (PTM) e da eventuali differenze di struttura primarie, come clip o varianti di sequenza. La natura relativamente semplice e ad alta produttività di queste misure le rende interessanti per la caratterizzazione e come controlli di monitoraggio in-process 7,8. L’analisi dei dati per questi esperimenti di solito richiede all’utente di definire lo spazio di ricerca per le forme molecolari (gamma di PTM o altre forme molecolari). Per le proteine glicosilate, questo spazio di ricerca è in gran parte guidato dall’eterogeneità dei glicoformi. Le combinazioni di più PTM, le configurazioni del legame disolfuro e altre variazioni lungo la struttura primaria rendono il calcolo di tutte le possibili forme molecolari un compito noioso. Pertanto, il calcolo manuale delle possibili forme molecolari è un processo che richiede tempo e risorse con un alto potenziale di errore umano.
Qui presentiamo uno strumento di calcolo della massa che è stato sviluppato considerando le caratteristiche più importanti delle molecole bioterapeutiche, come mAbs, bsAbs, ADC, ecc. Lo strumento consente di incorporare facilmente le variabili dello spazio di ricerca per il calcolo coerente delle masse e delle composizioni elementari. La natura modulare di questo strumento consentirà di svilupparlo ulteriormente e di applicarlo al calcolo della massa e alla corrispondenza della massa per altre modalità.
Il modulo GUI consente all’utente di specificare l’input per il calcolo della massa, come mostrato in Figura 1; In particolare, l’utente inserisce sequenze di amminoacidi di una sola lettera per catene di anticorpi leggeri e pesanti. Le modifiche comuni per la ciclizzazione N-terminale a catena pesante e il clipping della lisina con terminale C sono incluse come caselle di controllo. Inoltre, la formula chimica/composizione elementare può essere aggiunta/sottratta da queste catene proteiche attraverso la rispettiva casella di testo Chem Mod . Ciò consente all’utente la flessibilità di aggiungere una composizione elementare che include più modifiche post-traduzionali o un carico utile di piccole molecole nel caso di un ADC. Poiché la maggior parte degli anticorpi monoclonali terapeutici sono progettati per rimuovere i siti di glicosilazione nella catena leggera, la glicosilazione nella catena leggera è lasciata facoltativa e può essere specificata utilizzando una casella di controllo sulla GUI.
Una variante tipica dell’analisi della massa intatta per gli anticorpi è un’analisi di massa a subunità ridotte, in cui la catena leggera viene staccata dalla catena pesante riducendo i legami disolfuro intercatena. A seconda della forza dell’agente riducente utilizzato, i legami disolfuro intracatena possono essere scissi o meno. Gli utenti hanno la flessibilità di inserire il numero totale di legami disolfuro a seconda del sottotipo di IgG o nel caso di un ADC9 coniugato con cisteina.
L’applicazione calcola le masse in modo bottom-up, in cui le composizioni elementari vengono prima calcolate per le singole catene pesanti e catene leggere. Successivamente, la ciclizzazione N-terminale della catena pesante (HC) Lys-clipping viene presa in considerazione regolando le composizioni elementari calcolate. Eventuali modifiche chimiche specificate vengono quindi applicate alle catene pesanti e/o leggere. A seconda del tipo di analisi e dei modelli di legame disolfuro specificati dall’utente, il numero di idrogeni viene regolato per le due catene polipeptidiche. Le masse glicosilate HC e catene leggere (LC) (facoltative) vengono calcolate in base all’input dell’utente. Infine, vengono combinate più masse HC e LC e i numeri di legame disolfuro vengono aggiornati automaticamente per il calcolo della massa intatta.
Con molecole più grandi come le proteine intatte, le masse monoisotopiche non possono essere misurate a causa del difetto di massa additivo quando si utilizzano spettrometri di massa con il tipico potere risolutivo. Invece, le masse nominali o medie sono misurate o riportate 5,10,11,12,13. Le masse elementari medie possono variare in base alla fonte utilizzata per le masse curate14,15. Sebbene le differenze nelle masse elementari possano essere piccole, possono sommarsi a valori significativi per i calcoli del peso molecolare di grandi molecole. Le masse elementari medie utilizzate per impostazione predefinita nell’applicazione software sono mostrate nella Tabella supplementare 1. Per gli ambienti regolamentati come il campo della ricerca e sviluppo biofarmaceutico (R&S), è importante mantenere masse molecolari coerenti perché le variazioni delle masse possono implicare modifiche all’entità molecolare durante i depositi normativi. Per garantire la coerenza nell’uso delle masse elementari, un dizionario delle masse elementari è incluso nello strumento software come file di testo con valori separati da virgole (csv): Element_Mass.csv (Supplementary Coding File 1). Allo stesso modo, è incluso un elenco curato di composizioni di glicani tipicamente viste sugli anticorpi monoclonali: Glycan.csv (Supplementary Coding File 2). Entrambi i file vengono salvati nella stessa cartella di un’applicazione eseguibile e possono essere modificati dall’utente per utilizzare uno specifico elenco di masse elementari o una libreria di glicani.
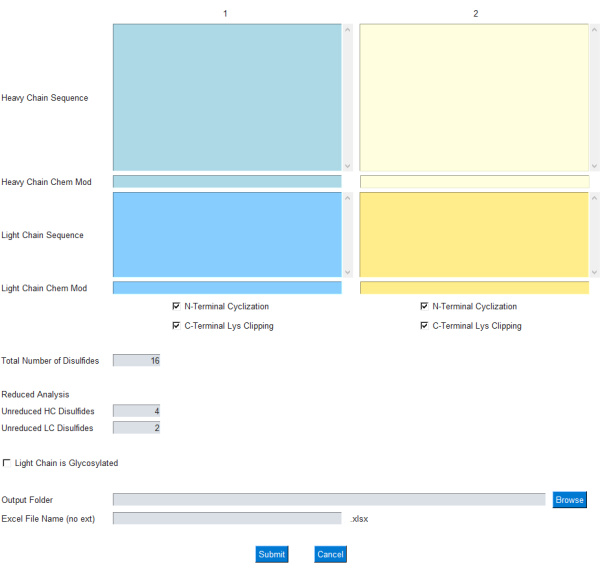
Figura 1: Interfaccia GUI per l’applicazione mAbScale. Il modulo GUI consente all’utente di specificare l’input per il calcolo della massa. L’utente inserisce sequenze di amminoacidi di una sola lettera per le catene di anticorpi leggeri e pesanti. Le modifiche comuni per la ciclizzazione N-terminale a catena pesante e il clipping della lisina C-terminale sono incluse come caselle di controllo. Le formule chimiche/composizioni elementari possono essere aggiunte/sottratte tramite la rispettiva casella di testo Chem Mod . Fare clic qui per visualizzare una versione più grande di questa figura.
