За последние два десятилетия биотерапевтические препараты превратились в основу современной фармацевтической промышленности. Пандемия SARS-CoV2 и другие опасные для жизни состояния еще больше увеличили потребность в более быстрой и широкой разработке биофармацевтических молекул 1,2,3.
Биотерапевтическая молекулярная масса имеет решающее значение для идентификации молекулы в сочетании с другими аналитическими анализами. Неповрежденные и уменьшенные массы субъединиц используются на протяжении всего жизненного цикла открытия и разработки в рамках стратегий контроля, направленных на поддержание качества, как описано в QTPP (Quality Target Product Profile)4.
Аналитические разработки в биофармацевтической промышленности в значительной степени опираются на измерения массы для анализа интактной массы и глубокой характеристики с использованием пептидного картирования или мониторинга многоатрибутного метода (MAM). В основе этих методов, использующих современные платформы масс-спектрометрии (МС), лежит возможность обеспечения точных измерений массы (ЧСС/АМ) с высоким разрешением. Большинство приборов ЧСС/АМ дают точность измерения массы в диапазоне 0,5-5 ppm, которая зависит от диапазона масс. Возможность точного измерения масс неповрежденных больших молекул позволяет быстро и уверенно идентифицировать крупномолекулярные терапевтические средства. Поскольку изотопное разрешение не может быть достигнуто в типичных экспериментальных условиях для больших молекул (>10 кДа), для сравнения и идентификации необходимо рассчитать средние массы 5,6.
Типичный масс-спектр интактного или субъединичного белка представляет собой общий профиль протеоформы, который содержит сводную информацию о различных молекулярных формах, возникающих в результате посттрансляционных модификаций (ПТМ), и любых первичных структурных различиях, таких как клипсы или варианты последовательностей. Относительная простота и высокая производительность этих измерений делают их привлекательными для определения характеристик и в качестве средств контроля в процессе производства 7,8. Анализ данных для этих экспериментов обычно требует от пользователя определения пространства поиска молекулярных форм (диапазона ПТМ или других молекулярных форм). Для гликозилированных белков это пространство поиска в значительной степени обусловлено гетерогенностью гликоформа. Комбинации нескольких ПТМ, конфигураций дисульфидных связей и других вариаций вдоль первичной структуры делают расчет всех возможных молекулярных форм утомительной задачей. Поэтому ручной расчет возможных молекулярных форм является трудоемким и ресурсоемким процессом с высокой вероятностью человеческой ошибки.
Здесь мы представляем инструмент расчета массы, который был разработан с учетом наиболее важных характеристик биотерапевтических молекул, таких как мАТ, бсАБ, АЦП и т.д. Инструмент позволяет легко вводить переменные пространства поиска для согласованного вычисления масс и элементного состава. Модульный характер этого инструмента позволит в дальнейшем развивать его и применять для вычисления массы и сопоставления масс для других модальностей.
Модуль GUI позволяет пользователю указать входные данные для расчета массы, как показано на рисунке 1; В частности, пользователь вводит однобуквенные аминокислотные последовательности для легких и тяжелых цепочек антител. Общие модификации для циклизации N-конца с тяжелой цепью и клипирования лизина C-концевой включены в качестве флажков. Кроме того, химическая формула/элементный состав могут быть добавлены/вычтены из этих белковых цепей через соответствующее текстовое поле Chem Mod . Это позволяет пользователю гибко добавлять элементный состав, который включает в себя несколько посттрансляционных модификаций или полезную нагрузку из малых молекул в случае АЦП. Поскольку большинство терапевтических мАТ спроектированы таким образом, чтобы удалять сайты гликозилирования в легкой цепи, гликозилирование в легкой цепи остается необязательным и может быть задано с помощью флажка в графическом интерфейсе.
Типичным вариантом анализа интактной массы на антитела является анализ приведенной субъединичной массы, при котором легкая цепь отделяется от тяжелой цепи путем уменьшения межцепочечных дисульфидных связей. В зависимости от силы используемого восстановителя внутрицепочечные дисульфидные связи могут расщепляться или не расщепляться. Пользователи могут вводить общее количество дисульфидных связей в зависимости от подтипа IgG или в случае цистеин-конъюгированного АЦП9.
Приложение рассчитывает массы по принципу «снизу вверх», при этом сначала рассчитываются элементные составы для отдельных тяжелых и легких цепей. Далее N-концевая циклизация тяжелой цепи (HC) Lys-клиппинг учитывается путем корректировки рассчитанного элементного состава. Любые указанные химические модификации затем наносятся на тяжелые и/или легкие цепи. В зависимости от типа анализа и характера дисульфидной связи, заданного пользователем, количество водорода корректируется для двух полипептидных цепей. Массы гликозилированного HC и легкой цепи (LC) (опционально) рассчитываются на основе вводимых пользователем данных. Наконец, несколько масс HC и LC объединяются, и номера дисульфидных связей автоматически обновляются для расчета неповрежденной массы.
С более крупными молекулами, такими как интактные белки, моноизотопные массы не могут быть измерены из-за дефекта аддитивной массы при использовании масс-спектрометров с типичной разрешающей способностью. Вместо этого измеряются номинальные или средние массы 5,10,11,12,13. Средние массы элементов могут варьироваться в зависимости от источника, используемого для курируемых масс14,15. Хотя различия в массах элементов могут быть небольшими, они могут складываться в значительные значения для расчетов молекулярной массы больших молекул. Средние массы элементов, используемые по умолчанию в программном приложении, приведены в Дополнительной таблице 1. Для регулируемых сред, таких как биофармацевтические исследования и разработки (НИОКР), важно поддерживать постоянную молекулярную массу, поскольку изменения масс могут повлечь за собой изменения молекулярной сущности во время подачи заявок в регулирующие органы. Чтобы обеспечить единообразие в использовании элементарных масс, словарь элементарных масс включен в программный инструмент в виде текстового файла с разделителями-запятыми (csv): Element_Mass.csv (Дополнительный файл кодирования 1). Аналогичным образом, включен курируемый список гликановых композиций, обычно встречающихся на мАТ: Glycan.csv (Дополнительный файл кодирования 2). Оба файла сохраняются в той же папке, что и исполняемое приложение, и могут быть изменены пользователем для использования определенного списка элементных масс или библиотеки гликанов.
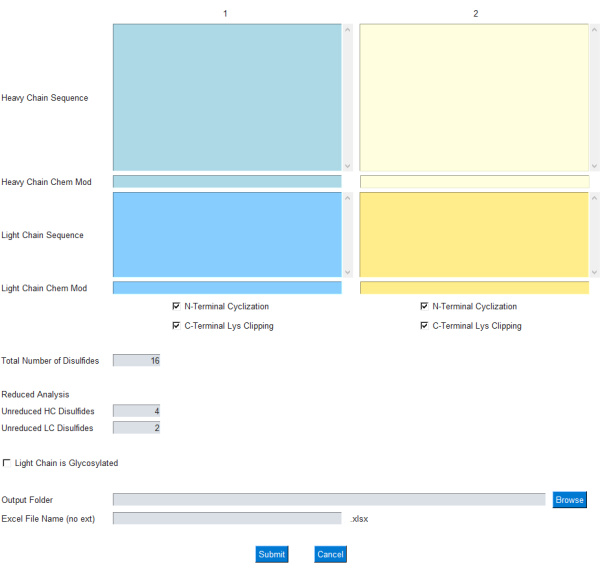
Рисунок 1: Графический интерфейс для приложения mAbScale. Модуль GUI позволяет пользователю указать входные данные для расчета массы. Пользователь вводит однобуквенные аминокислотные последовательности для легких и тяжелых цепочек антител. Общие модификации для циклизации N-конца тяжелой цепи и клипирования лизина C-концевой включены в качестве флажков. Химические формулы/элементные составы могут быть добавлены/вычтены через соответствующее текстовое поле Chem Mod . Пожалуйста, нажмите здесь, чтобы увидеть увеличенную версию этого рисунка.
