I løpet av de siste to tiårene har bioterapi utviklet seg til å bli en bærebjelke i den moderne farmasøytiske industrien. SARS-CoV2-pandemien og andre livstruende tilstander har ytterligere økt behovet for raskere og bredere utvikling av biofarmasøytiske molekyler 1,2,3.
Den bioterapeutiske molekylvekten er kritisk for identifisering av molekylet, i kombinasjon med andre analytiske analyser. De intakte og reduserte underenhetsmassene brukes gjennom hele oppdagelses- og utviklingslivssyklusen som en del av kontrollstrategier som tar sikte på å opprettholde kvaliteten, som beskrevet i QTPP (Quality Target Product Profile)4.
Analytisk utvikling i biofarmasøytisk industri er avhengig av massemålinger for intakt masseanalyse og dyp karakterisering ved hjelp av peptidkartlegging eller multiattributtmetode (MAM) overvåking. I sentrum av disse teknikkene som benytter moderne massespektrometri (MS) plattformer er evnen til å gi høyoppløselige nøyaktige masse (HR / AM) målinger. De fleste HR / AM-instrumenter gir massenøyaktigheter i området 0,5-5 ppm, som skalerer med masseområdet. Evnen til å måle masser nøyaktig for intakte store molekyler muliggjør rask og trygg identifisering av stormolekylære terapier. Siden isotopoppløsning ikke kan oppnås ved bruk av typiske eksperimentelle betingelser for store molekyler (>10 kDa), må gjennomsnittsmasser beregnes for sammenligning og identifisering 5,6.
Et typisk intakt eller subenhetsproteinmassespektrum representerer den overordnede proteoformprofilen, som inneholder sammensatt informasjon om de forskjellige molekylære formene som følge av posttranslasjonelle modifikasjoner (PTM) og eventuelle primære strukturforskjeller, for eksempel klipp eller sekvensvarianter. Den relativt enkle og høye gjennomstrømningen av disse målingene gjør dem attraktive for karakterisering og som overvåkingskontroller i prosessen 7,8. Dataanalyse for disse eksperimentene krever vanligvis at brukeren definerer søkeområdet for molekylære former (utvalg av PTM eller andre molekylære former). For glykosylerte proteiner er dette søkeområdet i stor grad drevet av glykoformheterogenitet. Kombinasjoner av flere PTM-er, disulfidbindingskonfigurasjoner og andre variasjoner langs primærstrukturen gjør beregning av alle mulige molekylære former til en kjedelig oppgave. Derfor er manuell beregning av mulige molekylære former en tid- og ressurskrevende prosess med høyt potensial for menneskelige feil.
Her presenterer vi et masseberegningsverktøy som ble utviklet med tanke på de viktigste egenskapene til bioterapeutiske molekyler, som mAbs, bsAbs, ADC, etc. Verktøyet gjør det enkelt å innlemme søkeromvariabler for konsistent beregning av masser og elementære komposisjoner. Den modulære naturen til dette verktøyet vil gjøre det mulig å videreutvikle og bruke til masseberegning og massematching for andre modaliteter.
GUI-modulen lar brukeren spesifisere inngangen for masseberegningen, som vist i figur 1; Spesielt går brukeren inn i aminosyresekvenser med én bokstav for lette og tunge antistoffkjeder. Vanlige modifikasjoner for tungkjedet N-terminal syklisering og C-terminal lysinklipping er inkludert som avmerkingsbokser. Videre kan den kjemiske formelen / elementsammensetningen legges til / trekkes fra disse proteinkjedene gjennom den respektive Chem Mod-tekstboksen . Dette gir brukeren fleksibiliteten til å legge til en elementær sammensetning som inkluderer flere posttranslasjonelle modifikasjoner eller en nyttelast med lite molekyl i tilfelle av en ADC. Siden de fleste terapeutiske mAbs er konstruert for å fjerne glykosyleringsstedene i lyskjeden, er glykosylering i lyskjeden valgfri og kan spesifiseres ved hjelp av en avmerkingsboks på GUI.
En typisk variasjon på intakt masseanalyse for antistoffer er en redusert subenhetsmasseanalyse, hvor den lette kjeden løsnes fra den tunge kjeden ved å redusere disulfidbindingene mellom kjedene. Avhengig av styrken av reduksjonsmidlet som brukes, kan de intrakjede disulfidbindingene spaltes eller ikke. Brukerne har fleksibilitet til å angi totalt antall disulfidbindinger avhengig av IgG-subtypen eller i tilfelle en cysteinkonjugert ADC9.
Applikasjonen beregner masser nedenfra og opp, der elementsammensetningene først beregnes for de enkelte tunge kjeder og lette kjeder. Deretter regnskapsføres tungkjede (HC) N-terminal syklisering Lys-klipping ved å justere de beregnede elementære sammensetningene. Eventuelle spesifiserte kjemiske modifikasjoner blir deretter påført de tunge og / eller lette kjedene. Avhengig av typen analyse og disulfidbindingsmønstrene spesifisert av brukeren, justeres antall hydrogener for de to polypeptidkjedene. De glykosylerte HC- og lettkjedemassene (LC) (valgfritt) beregnes basert på brukerens inndata. Til slutt kombineres flere HC- og LC-masser, og disulfidbindingsnumrene oppdateres automatisk for intakt masseberegning.
Med større molekyler som intakte proteiner kan monoisotopiske masser ikke måles på grunn av additivmassedefekten ved bruk av massespektrometre med typisk oppløsningskraft. I stedet måles eller rapporteres nominelle eller gjennomsnittlige masser 5,10,11,12,13. De gjennomsnittlige elementmassene kan variere basert på kilden som brukes til de kuraterte massene14,15. Mens forskjellene i elementmasser kan være små, kan de legge opp til signifikante verdier for molekylvektberegninger med store molekyler. De gjennomsnittlige elementmassene som brukes som standard i programvaren, er vist i tilleggstabell 1. For regulerte miljøer som biofarmasøytisk forskning og utvikling (FoU) -feltet, er det viktig å opprettholde konsistente molekylmasser fordi endringer i massene kan innebære endringer i molekylenheten under regulatoriske innleveringer. For å muliggjøre konsistens i bruken av elementmasser, følger en ordbok med elementmasser med programvareverktøyet som en kommaseparert tekstfil (csv): Element_Mass.csv (Supplementary Coding File 1). På samme måte er en kuratert liste over glykankomposisjoner som vanligvis ses på mAbs inkludert: Glykan.csv (Supplementary Coding File 2). Begge filene lagres på samme mappeplassering som et kjørbart program og kan endres av brukeren for å bruke en bestemt elementær masseliste eller glykanbibliotek.
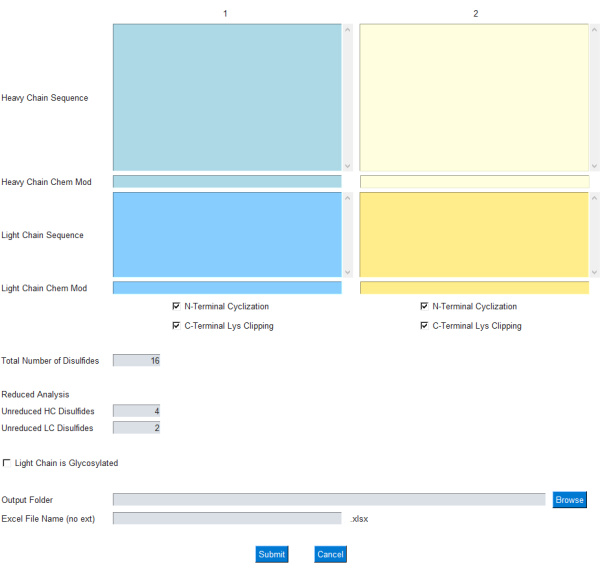
Figur 1: GUI-grensesnitt for mAbScale-programmet. GUI-modulen lar brukeren spesifisere inngangen for masseberegningen. Brukeren legger inn aminosyresekvenser på én bokstav for de lette og tunge antistoffkjedene. Vanlige modifikasjoner for tungkjedet N-terminal syklisering og C-terminal lysinklipping er inkludert som avmerkingsbokser. Kjemiske formler / elementære komposisjoner kan legges til / trekkes fra gjennom den respektive Chem Mod-tekstboksen . Klikk her for å se en større versjon av denne figuren.
