Growing Neural Stem Cells from Conventional and Nonconventional Regions of the Adult Rodent Brain
Summary
Neural stem cells harvested from the adult brain are increasing utilized in applications ranging from the basic research of nervous system development to exploring potential clinical applications in regenerative medicine. This makes rigorous control in the isolation and culturing conditions used to grow these cells critical to sound experimental outcomes.
Abstract
Recent work demonstrates that central nervous system (CNS) regeneration and tumorigenesis involves populations of stem cells (SCs) resident within the adult brain. However, the mechanisms these normally quiescent cells employ to ensure proper functioning of neural networks, as well as their role in recovery from injury and mitigation of neurodegenerative processes are little understood. These cells reside in regions referred to as "niches" that provide a sustaining environment involving modulatory signals from both the vascular and immune systems. The isolation, maintenance, and differentiation of CNS SCs under defined culture conditions which exclude unknown factors, makes them accessible to treatment by pharmacological or genetic means, thus providing insight into their in vivo behavior. Here we offer detailed information on the methods for generating cultures of CNS SCs from distinct regions of the adult brain and approaches to assess their differentiation potential into neurons, astrocytes, and oligodendrocytes in vitro. This technique yields a homogeneous cell population as a monolayer culture that can be visualized to study individual SCs and their progeny. Furthermore, it can be applied across different animal model systems and clinical samples, being used previously to predict regenerative responses in the damaged adult nervous system.
Introduction
The central dogma of neurobiology, laid down by the fundamental observations of the brain cytoarchitecture made by Ramón Y. Cajal over a century ago, held that neurogenesis was unlikely after adolescence given the complexity of the neural networks found in the CNS1. Despite the work of Altman in the 1960s, and later Kaplan, demonstrating that 3H-thymidine could be found in mature neurons indicating that in fact neurons were being generated in distinct areas of the adult brain, the dogma continued to hold2,3. Evidence continued to mount with Nottebohm’s research describing the seasonal changes in the number of neurons present in songbird brains4. It wasn’t until 1999, when Gould et al. published work on the generation of neurons in the hippocampus increases with the performance of associative learning tasks in rat, as well as the observations of Kornack and Rakic demonstrating continued neurogenesis in the adult macaque that the concept of a less rigid, more plastic brain, was recognized5,6.
The search for the cellular source for these de novo generated neurons lead to the discovery of a discrete population of stem cells (SCs) that reside in areas of the brain referred to as niches7. The subventricular zone and sub granular zone of the hippocampus are considered to be the two principal neurogenic regions8,9. Cells isolated from these locations display the classic characteristic of embryonic or fetal derived SCs, self-renewal and differentiation potential. In the case of neural stem cells (NSCs), they can be differentiated into neurons, astrocytes, and oligodendrocytes. In addition, these SCs stain positive for fetal NSC markers such as the intermediate filament protein nestin10. More recent work highlights that SCs might not be limited to these two areas, and are in fact localized throughout the brain as a largely quiescent population of cells tightly associated with the vasculature11.
The observations that SCs are mobilized in response to injury suggests the possibility of being able to utilize these cells for regenerative purposes to aid in the recovery from neurodegenerative disorder and stroke12,13. This is not unlike the role that mesenchymal stem cells (MSCs) play in the healing of connective tissues, which are found as perivascular cells that have the potential to become osteoblasts, chondrocytes, and adipose cells14. However, NSCs cannot be harvested in the same manner as MSCs from bone marrow by routine aspiration and density gradient centrifugation techniques and subsequently utilized in autologous cell based therapies. As a consequence, other sources of cells, such as the use of fetal NSCs or neuronal precursors derived from embryonic stem cells have been extensively explored in animal disease and injury models with varying degrees of success15. Induced pluripotent stem cell technologies utilizing somatic cell sources offer another potential avenue for producing therapeutically useful cell-based therapies for a wide range of applications, overcoming the limited availability and ethical concerns regarding the use of embryonic cells and fetal tissues16. However, clinical translation of these findings has proven to be a difficult task, as demonstrated in the struggles of treating various neurological conditions with SC-based therapeutic approaches17,18, as well as a tortuous path to regulatory clearance. As an alternative approach, introduction of specific pharmacological treatments can modulate NSC numbers and facilitate recovery in models of Parkinson’s disease and stroke19. Whatever the strategy might be, understanding how to effectively manipulate these cells requires an accessible in vitro system.
Cultures of NSCs can be performed either as aggregate cultures, also known as neurospheres, or as a monolayer8,20. Both techniques have been widely used, allowing for the establishment of defined culture conditions, i.e. use of Epidermal Growth Factor (EGF) or basic Fibroblast Growth Factor (bFGF) as a mitogen source, that provide for the expansion of multipotent precursors. While neurosphere cultures may be better suited for studying clonal propagation capability of an isolated cell type, the system has been shown to produce a mixed population of cells during expansion21. In addition, the closed structure of neurospheres makes pharmacological manipulation of the cells impractical, and the interpretation of the influence these factors may have could be confounded due to the microenvironment established within the neurosphere itself. Monolayer cultures, on the other hand, can be employed in high throughput screens of small molecule libraries, providing a powerful tool to explore the signal transduction mechanisms that regulate SC growth and differentiation and opens the opportunity to discover novel compounds that specifically target this cell population.
As a consequence, the ability to reproducibly generate cultures of adult NSCs from different regions of interest in the brain can be used in a broad spectrum of research applications, ranging from developmental studies of the central nervous system (CNS) to exploring novel regenerative medicine approaches. The protocol presented here demonstrates how to dissect and assess the differentiation potential of CNS SCs isolated from the adult rodent brain.
Protocol
The work adheres to the Declaration of Helsinski and the ARVO Animal Statement. Animals were used for tissue collection and all relevant methods were followed according to the instructions of the animal facility at the University of Dresden. Animals were handled and housed according to the German Federal guidelines for the use and care of laboratory animals, and the study was approved by the Landesdirektion Dresden. Please consult with your institution’s veterinary policy (IACUC or other board) regarding appropriate euthanasia methodology.
Specific stock and working concentrations of reagents can be found in the Reagent Tables accompanying this protocol.
1. Culture Dish Preparation
- Coat dishes with a sufficient volume of 75 μg/ml poly-L-ornithine (i.e. 2 ml/well of a 6-well plate) overnight in the cell culture incubator 2 days prior to the scheduled dissection date.
- Aspirate the poly-L-ornithine the following day and wash each well 2x with 3 ml of PBS for 5 min.
- Remove the final wash, then add fibronectin (diluted 1:250, 4 μg/ml in PBS) to the plate and place the dishes in the incubator overnight.
- Aspirate the fibronectin on the day of dissection and wash the dishes 2x with PBS. Return the plates still containing the final PBS wash to the incubator until the dissociated tissue is ready for culturing. At that time, remove the PBS prior to plating the tissue.
- Store plates coated with poly-L-ornithinein the incubator for up to 3 weeks. Plates coated with fibronectin can be stored for up to 1 week. Fibronectin coating can be done in as little as 2 hr if urgent. Fibronectin is susceptible to denaturation; do not agitate the solution or let dishes dry.
2. Dissection/Plating
- This protocol applies to the use of 3 adult rats (3-6 months of age).
- Chill the brain sectioning block on ice prior to euthanizing the rat.
- Place a 15 ml Falcon tube that contains 5 ml of N2 medium with added bFGF on ice.
- Euthanize the rats according to your institution’s veterinary policy.
- Remove the brain from the skull by making an incision down the midline of the head. Use forceps to peel away the bone allowing for the brain to be carefully extracted.
- Place the brain into a 10 cm Petri dish containing ice cold PBS. This will remove any residual hair and blood. (Figure 1A)
- Place the brain into the chilled sectioning block. (Figure 1B)
- Insert one new clean razor blade into the block as pictured in Figure 1C.
- Insert a second clean razor blade 3 mm caudal to the first as illustrated in Figure 1D.
- Carefully lift the blades from the block, taking with them the section of interest.
- Float the section off the blade by immersing it in PBS. (Figure 1E)
- Harvest tissue from the area of interest (in this example we used the anterior commissure, (anterior) ACA, Figure 1F) using #5 forceps and collect into a 15 ml conical tube containing 1 ml N2 medium containing bFGF.
- Move the tube into a laminar flow cell culture hood, continuing the remainder of the protocol under sterile conditions.
- Mechanically dissociate the tissue using a 1 ml pipette tip attached to a P1000 pipettor. Pipette up and down several times (approximately 20x at a rate of 1 pipetting cycle per second) while pressing the opening of the pipette tip against the bottom of the conical tube (Figures 1G 1-3). Stop trituration when the solution becomes homogeneous.
- Allow the solution to sit for 2 min to allow any larger nondissociated tissue to settle to the bottom. (Figure 1G 4)
- Collect the homogeneous supernatant and place into a conical tube containing 8.5 ml of N2 media plus 20 ng/ml bFGF, 500 ng/ml Dll4, 500 ng/ml Ang2, and 200 nMJAK Inhibitor.
- Plate them into 3 wells of a 6-well plate using 3 ml of diluted cell solution/well.
- Place the plate into a humidified cell incubator at 37 °C, 5% CO2, 5% O2.
3. Daily Care
- Replace the medium on the culture dishes with N2 Medium containing bFGF, Ang2, Dll4, and JAK inhibitor 24 hr after plating.
- Add a bolus of bFGF, Dll4, Ang2, and JAK inhibitor on the next day.
- Continue this alternating process of media changes and factor additions for a total of 10-14 days.
4. Induction of Differentiation
- Withdraw bFGF, Dll4, Ang2, and JAK inhibitor containing medium and replace it with N2 medium that does not contain any additional factors.
- Change the medium every second day.
- Fix the cells after 10 days and perform immunocytochemistry.
5. Immunocytochemical Detection
- Aspirate the medium and fix cell culture plates with 2 ml of 4% paraformaldehyde for 20 min.
- Wash 2x with 3 ml of PBS for 5 min each.
- Aspirate PBS, then permeabilize and block the cells by adding 2 ml of PBS containing 5% normal donkey serum (NDS) and 0.1% Triton X-100 for 20 min.
- Prepare the primary antibody mix in PBS containing 5% NDS. Anti-nestin antibody can be used for identifying SCs, while TuJ1 (class III β-tubulin), GFAP, and CNPase antibodies can be used together for a triple staining of differentiation markers.
- Aspirate the blocking solution and add 1.5 ml of the primary antibody mixture to each well. Incubate at room temperature for 90 min.
- Remove the primary antibodies and wash 2x with 3 ml of PBS, and 1x with 3 ml of PBS containing 5% NDS for 5 min each.
- Prepare the secondary antibodies in PBS containing 5% NDS.
- Aspirate the final wash and add 1.5 ml of secondary antibody solution to each well. Incubate in the dark for 40 min at room temperature.
- Remove the secondary antibody solution and add 2 ml of a freshly prepared DAPI stain (500 ng/ml in PBS). Incubate for 3 min.
- Aspirate the DAPI solution then wash 3x with 3 ml of PBS for 5 min each. The cells can now be visualized with a fluorescence microscope.
Representative Results
Identifying the region of interest from which to harvest neural stem cells is the critical first step and will define the amount of time it takes to get a confluent plate of cells. For example, the SVZ is a classic neurogenic area and therefore has a higher relative proportion of neural stem cells. However, the technique presented here can be used with other regions not often recognized as having a robust neurogenic potential during adulthood. For the purposes of this protocol, we used the anterior commissure (anterior part). While some of the cells and debris will settle upon plating, the medium will normally remain turbid as a consequence of the amount of cellular material that is present following trituration. The first medium change at 24 hr will remove the majority of this. As visualized by light microscopy, there will appear to be a large amount of cellular material remaining, along with blood vessels, even after the first medium change. Over the course of the next few days, colonies of a distinct proliferating cell population will become visible under the microscope (Figure 2A). These are the neural stem cells that have been selected for growth by the use of N2 medium containing bFGF, Ang2, Dll4, and JAK inhibitor. In addition, a more elongated spindle-like cell population may also be present (Figures 2B and C).These are not neural stem cells (likely radial glia based upon morphology and staining), they will eventually be overtaken by the more rapid proliferating neural stem cell population. Over the course of a week to 14 days, the cells will become more dense until they reach confluence (Figure 2D). These cultures stain positive for a number of stem cell markers, including Nestin, Hes3, and Sox2. (Figures 2E-H), thus providing confidence that the cell populations that have been selected and expanded are indeed neural stem cells. Further confirmation comes from the ability to withdrawal FGF from the medium, allowing the cells to differentiate for 10 days to generate neurons, astrocytes, and oligodendrocytes (Figure 2I).
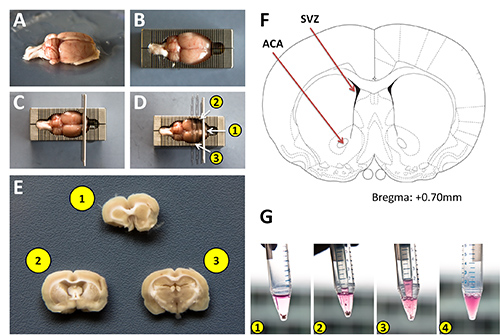
Figure 1: Isolation of rat brain sections for harvesting adult neural stem cells. A) Adult rat brain after washing with PBS. B) Rat brain placed into a chilled stainless steel sectioning block. C) Approximate positioning of the initial razor blades within the block to produce coronal sections the brain. D) Placement of 2 additional razor blades into the sectioning block. Note, thinner double edged blades were used for demonstration purposes to allow for easier visualization of the sections in the block. E) Coronal brain sections containing areas to be harvested. F) Schematic diagram from the Paxinos brain atlas highlighting the subventricular zone as a classic neurogenic region, and the anterior commissure from which cells were harvested for this protocol. G) Sequential images of the tissue dissociation process using a 1 ml pipette tip attached to a P1000 micropipettor. Click here to view larger image.
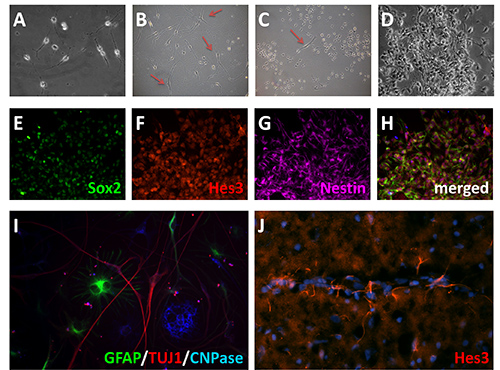
Figure 2: Adult neural stem cells in vitro. A) Adult neural stem cell culture after one week. B) Examples of a more broad spindle morphology cells (red arrows) occasionally present in the cultures that are not neural stem cells. C, D) Under the culture conditions defined in this protocol, the neural stem cells preferentially expand. E-H) Expression of neural stem cell markers following 14 days in vitro, Sox2 (green)(E), Hes 3 (red)(F), and Nestin (violet)(G). I) Differentiation of neural stem cells into neurons, astrocytes, and oligodendrocytes. Following mitogen withdrawal, cells were allowed to differentiate for 10 days, then immunostained for GFAP (green), TuJ1 (red), and CNPase (blue). J) Hes3 immunostaining in adult rat brain. Click here to view larger image.
Discussion
Many of the steps critical to successful isolation, expansion, and differentiation of neural stem cells from the adult brain are shared in common with standard tissue culture techniques. The objective to bear in mind is that NSCs isolated from the adult brain should maintain as many of their in vivo characteristics as possible (Figure 2J). Often a balance between necessary caution and appropriate speed needs to be struck. Care with the isolation of the brain from the skull will make the identification of the region of interest significantly easier. Removing the tissue as quickly as possible following animal euthanization, and keeping the brain cold during harvesting and washing keeps it more rigid and greatly assists in handling of the tissue when it is placed into the stainless steel sectioning mold. This also minimizes the potential for ripping of the tissue during the sectioning process. The protocol also benefits from being able to get the cells to the point of plating quickly, as this increases cell viability once they are placed in vitro. Proper tissue washing prior to sectioning also helps reduce the potential of contamination in the cultures. During the dissociation process, trituration with a 1 ml pipette tip should be vigorous, but done in a way as not to generate excess bubbles or frothing of the cell suspension which negatively impacts cell survival. The first medium change is also crucial as it removes the majority of the nonattached cells and a cellular debris that results from the tissue dissociation process which contains factors that are detrimental to the initial survival of the stem cell population. For improved visualization following immunostaining, the freshly isolated cells can be plated on 25 mm glass coverslips placed in the bottom of the 6-well plates. The concentrations of poly-L-ornithine and fibronectin used in coating should be increased to 500 μg/ml and 20 μg/ml, respectively. As an alternative, multiple smaller coverslips (i.e. 12 mm) can be placed into each well. These can then be processed for immunostaining independently in separate wells of a 24-well plate, scaling down the amounts of staining reagents used to take into account the smaller volumes. The coverslips are then mounted on slides using a standard aqueous mounting medium.
Defining the presence of NSCs in the adult brain is of major interest in the field of neurobiology. There are two general approaches to identify a stem cell. In one, a set of biomarkers based upon a gene expression profile of the stem cell population in question can be used to image them by immunohistochemistry. Although experimentally simple, it gives no functional data, nor defines whether the cells have the fundamental properties of stem cells, the ability to self renew or differentiate into multiple different cell types. In the other approach, cells are isolated in either a pure or heterogeneous population, placed in culture, and there self renewal and multipotential properties are assessed using the living cell population. This approach gives functional data, and can be used to unequivocally prove “stemness” in these particular in vitro experimental conditions. However, the difficulty with maintaining these cells in culture has hindered this second functional approach. In fact, the inability to grow NSCs from other areas of the brain has left the impression that there are only a few specialized niches where they reside.
The method presented here allows for the culturing of NSCs from many different areas of the CNS. This technique is based upon our work that elucidated a signal transduction pathway of great importance to controlling the number of NSCs both in vitro and in vivo. The factors included in this protocol were selected based upon extensive signal transduction studies, demonstrating that they promote NSC growth via a common transcription factor Hes3. Our previous studies show that this particular combination yielded an even greater increase in NSC numbers when compared to using them individually. The choice of which pharmacological support is added to the culture should be considered in the context of the biology being studied. For example, while both Ang2 and Dll4 have a positive impact on NSC growth, they have opposing effects on vasculature formation22,23. Through further optimizing of the culture conditions, additional novel biomarkers beyond Hes3 will likely be identified and expand the recognized number of NSCs in the adult brain even further. This is exemplified by our observations that distinctions among the Hes3+ cell population from different brain areas can be made. For example Hes3+ cells from the spinal cord, like those from the SVZ and ACA, show a several fold increase in their number when cultured with these factors. In addition, Hes3+ cells from all these regions can efficiently generate neurons, astrocytes, and oligodendrocytes. However, the morphology and gene expression of the differentiated cell types is not identical. Additional biomarkers that distinguish among different Hes3+ cell types will be a welcome addition to the field. The method presented here allowing for the efficient generation of NSC cultures can be used as a tool towards this goal.
Like the NSC markers nestin and Sox2, the transcription factor Hes3 identifies a cell population that upon isolation can be propagated in vitro as well as differentiated into the principal cell types that constitute the nervous system, neurons, astrocytes, and oligodendrocytes11. However, unlike these more commonly used markers, Hes3 also identifies quiescent NSCs; as a consequence, many Hes3+ cells do not incorporate indicators of mitosis (3H-thymidine or BrdU) under homeostatic conditions (and thus have avoided classic detection techniques). Application of the protocol described here was fundamental in the discovery of this NSC population. The number of these cells increases in culture when treated with factors produced from the vascular endothelium, including the Notch ligand Delta4, and Angiopoetin2, consistent with their perivascular localization in vivo24.
Having ready access to these cells isolated from the adult brain allows for experimentation to examine how the cells respond at the signal transduction level to various factors, and provides some predictive indications of how the cells will respond in vivo. Looking beyond regenerative medicine, we demonstrated that the culture conditions described here have relevance to cancer research as well. They better represent the environment that cancer stem cells isolated from Glioblastoma multiforme experience while in the patient, allowing for the study of signaling pathways that can be manipulated to oppose their growth30. The broad application of this technique could have significant implications for multiple aspects of research and medicine.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was funded (in part) by the Helmholtz Alliance ICEMED – Imaging and Curing Environmental Metabolic Diseases, through the Initiative and Network Fund of the Helmholtz Association, a grant from the Else Kroener-Fresenius Foundation, and a grant from the Deutsche Forschungsgemeinschaft (SFB 655: Cells into tissues).
Materials
| 6-well tissue culture dishes | BD Biosciences | 353934 | ||
| Poly-L-ornithine | Sigma-Aldrich | P-3655 |
5 mg/ml stock solution prepared in double distilled water (Stable for several months at -20 °C). Working concentration 0.5 mg/ml in water (Stable for 1 month at 4 °C). |
|
| Fibronectin | R&D Systems | 1030-Fn | Do not agitate stock solution | |
| Filtration Apparatus | Corning Life Sciences | 430769 | ||
| DMEM/F-12 | Mediatech | 10-090-CV | See note below for complete N2 media preparation | |
| Apo-transferrin | Sigma-Aldrich | T-2036 | ||
| Insulin | Sigma-Aldrich | I-0516 | ||
| Putrescine | Sigma-Aldrich | P-5780 | 1 M stock solution in ddH2O (Stable at 20 °C for 6 months) | |
| Sodium Selenite | Sigma-Aldrich | S-5261 | 500 µM stock solution in ddH2O (Stable at -20 °C for 6 months) | |
| Progesterone | Sigma-Aldrich | P-8783 | 100 µM stock solution in ethanol (Stable at -20°C for 6 months) | |
| Penicillin/Streptomycin | Invitrogen | 15140-122 | ||
| Phosphate Buffered Saline | Mediatech | 21-040-CV | ||
| Basic Fibroblast Growth Factor (bFGF) | R&D Systems | 233-FB | Working concentration 20 ng/ml | |
| Delta4 (Dll4) | R&D Systems | 1389-D4 | Working concentration 500 ng/ml | |
| Angiopoetin 2 (Ang2) | R&D Systems | 623-AN | Working concentration 500 ng/ml | |
| JAK Inhibitor | Calbiochem | 420099 | Working concentration 200 nM | |
| Bovine Serum Albumin | Sigma-Aldrich | A-2058 | ||
| 15- and 50-ml Conical tubes | Corning Life Sciences | 430053, 430829 | ||
| Other necessary items include: General dissection instruments, including razor blade and forceps, Adult rat (3-6 months old; Sprague-Dawley or Long Evans), CO2 intoxication chamber, Laminar flow hood for cell culture and incubator Incubator (humidified, 37 °C, 5% CO2, 5% O2). Note: For complete N2 media preparation, to one bottle of DMEM/F-12 (500 ml) add 0.05 g of apotransferin, 0.0125 g of insulin (freshly predissolved in 1 ml of 10 mM NaOH), 50 μl of putrescine, 30 μl of sodium selenite, 100 μl of progesterone stocks, and 5 ml of penicillin/streptomycin solution. Adjust pH to 7.2, if needed. Filter-sterilize and store at 4 °C for up to 3 weeks and protect from light. | ||||
| [header] | ||||
| Immunofluorescence Reagents Table | ||||
| Paraformaldehyde | Electron Microscopy Sciences | 15719 | ||
| Normal Donkey Serum (NDS) | Sigma-Aldrich | D-9663 | ||
| Triton X-100 | Sigma-Aldrich | T-8787 | ||
| 4,6-Diamidino-2-phenylindole (DAPI) | Sigma-Aldrich | D-8417 | 5 mg/ml stock solution in methanol | |
| [header] | ||||
| Primary Antibody Table | ||||
| Nestin | Chemicon | MAB353 |
Dilution Factor: 1:400 Species: Mouse IgG1 |
|
| Hes3 | Santa Cruz | sc-25393 |
Dilution Factor: 1:100 Species: Rabbit IgG |
|
| Sox2 | R&D Systems | MAB2018 |
Dilution Factor: 1:100 Species: Mouse IgG2a |
|
| CNPase | Chemicon | MAB326 |
Dilution Factor: 1:200 Species: Mouse IgG1 |
|
| β-tubulin III (TUJ1) | R&D Systems | MAB1195 |
Dilution Factor: 1:500 Species: Mouse IgG2a |
|
| Glial Fibrillary Acidic Protein (GFAP) | Dako North America | Z0334 |
Dilution Factor 1:500 Species: Rabbit |
|
| [header] | ||||
| Secondary Antibody Table | ||||
| Alexa 568 | Invitrogen | A-21124 |
Dilution Factor: 1:200 Species: Goat anti Mouse IgG1 |
|
| Alexa 488 | Invitrogen | A-21131 |
Dilution Factor: 1:200 Species: Goat anti Mouse IgG2a |
|
| Cy5 | Jackson ImmunoResearch | 59883 |
Dilution Factor: 1:200 Species: Goat anti Rabbit |
|
References
- Cajal, R. Y. . Comparative Study of Sensory Areas of the Human Cortex. , (1899).
- Altman, J., Das, G. D. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J. Comp. Neurol. 124, 319-335 (1965).
- Kaplan, M. S., Hinds, J. W. Neurogenesis in the adult rat: electron microscopic analysis of light radioautographs. Science. 197, 1092-1094 (1977).
- Nottebohm, F. Neuronal replacement in adulthood. Ann. N.Y. Acad. Sci. 457, 143-161 (1985).
- Gould, E., Reeves, A. J., Graziano, M. S., Gross, C. G. Neurogenesis in the neocortex of adult primates. Science. 286, 548-552 (1999).
- Kornack, D. R., Rakic, P. Continuation of neurogenesis in the hippocampus of the adult macaque monkey. Proc. Natl. Acad. Sci. U.S.A. 96, 5768-5773 (1999).
- Alvarez-Buylla, A., Kohwi, M., Nguyen, T. M., Merkle, F. T. The heterogeneity of adult neural stem cells and the emerging complexity of their niche. Cold Spring Harb. Symp. Quant. Biol. 73, 357-365 (2008).
- Reynolds, B. A., Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 255, 1707-1710 (1992).
- Luskin, M. B. Restricted proliferation and migration of postnatally generated neurons derived from the forebrain subventricular zone. Neuron. 11, 173-189 (1993).
- Lendahl, U., Zimmerman, L. B., McKay, R. D. CNS stem cells express a new class of intermediate filament protein. Cell. 60, 585-595 (1990).
- Androutsellis-Theotokis, A., et al. Targeting neural precursors in the adult brain rescues injured dopamine neurons. Proc. Natl. Acad. Sci. U.S.A. 106, 13570-13575 (2009).
- Jin, K., et al. Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc. Natl. Acad. Sci. U.S.A. 98, 4710-4715 (2001).
- Zhang, R. L., Zhang, Z. G., Chopp, M. Ischemic stroke and neurogenesis in the subventricular zone. Neuropharmacology. 55, 345-352 (2008).
- Caplan, A. I. Why are MSCs therapeutic? New data: new insight. J. Pathol. 217, 318-324 (2009).
- Goldman, S. Stem and progenitor cell-based therapy of the human central nervous system. Nat. Biotechnol. 23, 862-871 (2005).
- Sun, N., Longaker, M. T., Wu, J. C. Human iPS cell-based therapy: considerations before clinical applications. Cell Cycle. 9, 880-885 (2010).
- Amariglio, N., et al. Donor-derived brain tumor following neural stem cell transplantation in an ataxia telangiectasia patient. PLoS Med. 6, e1000029 (2009).
- Lindvall, O., Bjorklund, A. Cell therapeutics in Parkinson’s disease. Neurotherapeutics. 8, 539-548 (2011).
- Kittappa, R., Bornstein, S. R., Androutsellis-Theotokis, A. The Role of eNSCs in Neurodegenerative Disease. Mo.l Neurobiol. , (2012).
- Cattaneo, E., McKay, R. Identifying and manipulating neuronal stem cells. Trends Neurosci. 14, 338-340 (1991).
- Jensen, J. B., Parmar, M. Strengths and limitations of the neurosphere culture system. Mol. Neurobiol. 34, 153-161 (2006).
- Duarte, A., et al. Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev. 18, 2474-2478 (2004).
- Maisonpierre, P. C., et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 277, 55-60 (1997).
- Androutsellis-Theotokis, A., et al. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature. 442, 823-826 (2006).
- Alderson, R. F., Alterman, A. L., Barde, Y. A., Lindsay, R. M. Brain-derived neurotrophic factor increases survival and differentiated functions of rat septal cholinergic neurons in culture. Neuron. 5, 297-306 (1990).
- Johe, K. K., Hazel, T. G., Muller, T., Dugich-Djordjevic, M. M., McKay, R. D. Single factors direct the differentiation of stem cells from the fetal and adult central nervous system. Genes Dev. 10, 3129-3140 (1996).
- Ourednik, J., Ourednik, V., Lynch, W. P., Schachner, M., Snyder, E. Y. Neural stem cells display an inherent mechanism for rescuing dysfunctional neurons. Nat. Biotechnol. 20, 1103-1110 (2002).
- Ryu, J. K., Cho, T., Wang, Y. T., McLarnon, J. G. Neural progenitor cells attenuate inflammatory reactivity and neuronal loss in an animal model of inflamed AD brain. J. Neuroinflammation. 6, 39 (2009).
- Androutsellis-Theotokis, A., et al. Angiogenic factors stimulate growth of adult neural stem cells. PLoS One. 5, e9414 (2010).
- Park, D. M., et al. Hes3 regulates cell number in cultures from glioblastoma multiforme with stem cell characteristics. Sci. Rep. 3, 1095-1010 (2013).
- Poser, S. W., Shostak, S., et al. Ch. 5. Cancer Stem Cells – The Cutting. , 89-110 (2011).
- Androutsellis-Theotokis, A., Rueger, M. A., Mkhikian, H., Korb, E., McKay, R. D. Signaling pathways controlling neural stem cells slow progressive brain disease. Cold Spring Harb. Symp. Quant. Biol. 73, 403-410 (2008).
