QCM-D Analysis of Structured Woody Polymer Film Fabrication
The LbL adsorption of lignin, NFC and PDDA was monitored in real-time with QCM-D in two different experiments involving two types of lignins. This analysis method is very sensitive to detect changes in frequency when molecules adsorb to the surface of the quartz crystal. Figure 1 contains a detailed description of the QCM-D response in one deposition cycle, which involves two bilayers (PDDA:HMWL and PDDA:NC). The data represents the normalized change in frequency and dissipation of the 7th overtone (the instrument detects the fundamental frequency and the 3-13 odd harmonic overtones). A baseline was first obtained with pH 10.5 milli-Q water (termed as buffer), followed by introduction of the cationic polymer, PDDA. The introduction of this polymer (step 1) is associated with a decrease in ΔF, and a corresponding increase in ΔD. This response is attributed to a combination of adsorption of PDDA on the gold coated quartz substrate and the change in the bulk effects of the liquid in contact with the vibrating crystal. Step 1 was followed by a rinse step (step 2) with the buffer to remove the excess/unbound polymer, and to negate the frequency and dissipation response due to the bulk effects of the polymer solution. Therefore, a rinse was performed after each polymer adsorption step. The net change in ΔF and ΔD from the baseline after step 2 is due to the irreversible adsorption of PDDA. In step 3, the lignin solution was introduced, which resulted in a decrease in ΔF and a corresponding increase in ΔD. Step 4, the rinse step caused a slight increase in ΔF, however the ΔD remained unchanged, which suggests that lignin is deposited as a rigid layer over the PDDA layer when in contact with gold coated quartz substrate. To deposit the second bilayer, (PDDA:NC), the PDDA solution was reintroduced over the lignin layer (step 5). The introduction of PDDA solution was associated with a slight decrease in ΔF, and a significant increase in ΔD. However, after the initial drop, there was a gradual increase in ΔF followed by a plateau. After the buffer rinse (step 6), the net change in ΔF and ΔD after the deposition of PDDA on the lignin layer (ΔF = -31.6 Hz; ΔD = 1.3 x 10-6) was found to be slightly lower than the previous layer (ΔF = -33.2 Hz; ΔD = 1.7 x 10-6). This change is the result of a strong interaction between PDDA and lignin56,57, which may have caused partial desorption of loosely bound lignin deposited in step 3 (note in AFM section below, lignin remains in system). In step 7, the NC suspension was introduced on the PDDA layer resulting in an increase in ΔF and a corresponding decrease in ΔD. This change was found to be irreversible after the rinse step (step 8) suggesting that NC has been irreversibly deposited on PDDA. In the current study only four deposition cycles (8 bilayers, 4 cycles) were performed, because the ΔF and ΔD change beyond this number of cycles was found not to be reproducible. Figure 2 shows the normalized change in ΔF and ΔD of the 7th overtone as a result of the sequential adsorption of polymers PDDA, HMWL and NC after four deposition cycles. It should be noted that the adsorption of the polymers did not follow a linear change in ΔF and ΔD with the addition of each bilayer, which has also been noted with other LBL systems49,58. The LBL adsorption of NC, PDDA and OL (NC-PDDA-OL), shown in Figure 3, was found to follow the sequential adsorption process observed with NC-PDDA-HMWL. Nonetheless, the systems were found to differ with respect to the exact amount of the polymers deposited in each layer. The difference between these two systems is due to the type of the lignin used, as the cationic polymer and NC used were the same in both systems.
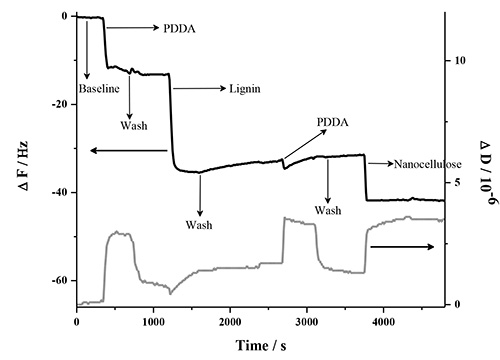
Figure 1. Description of the steps involved in the 1st deposition cycle of LBL adsorption of NC-PDDA-HMWL. The figure shows the normalized change in ΔF and ΔD of the 7th harmonic.
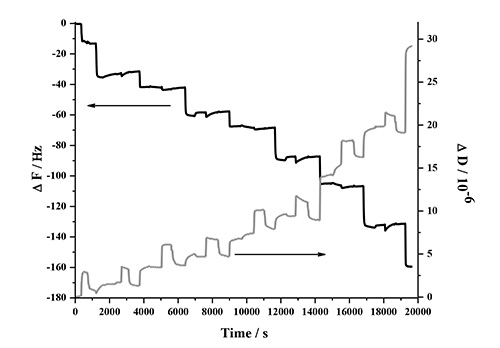
Figure 2. Frequency and dissipation response of the 7th harmonic as a result of the LBL adsorption of NC-PDDA-HMWL in 4 deposition cycles (8 bilayers).
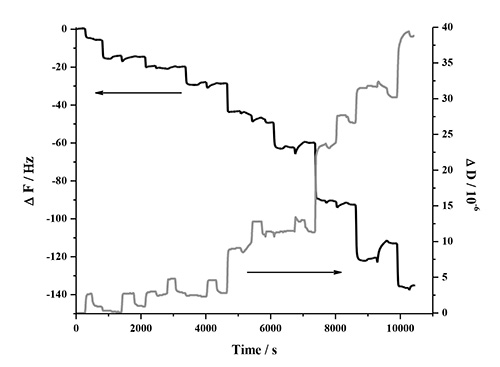
Figure 3. Frequency and dissipation response of the 7th harmonic as a result of the LBL adsorption of NC-PDDA-OL in 4 deposition cycles (8 bilayers).
Imaging Film Build-up with Atomic Force Microscopy
AFM images revealed complete coverage of the surface with lignin in the first PDDA-lignin bilayer for both HMWL and OL (Figure 4). The RMS roughness values for HMWL and OL were 1.6 and 3.8 nm, respectively. AFM images of the first PDDA-NC bilayer showed that the surface was not completely covered with NC fibrils, and revealed scattered NC fibrils in the 2.5 x 2.5 mm images (Figure 5a, RMS roughness 1.6 nm). However, as the layer build-up continued, more uniformity was found for the fibrils deposited, as seen in Figure 5b (RMS roughness 5.3 nm). Similar results were seen with QCM-D data, as ΔF for NC changed greater in magnitude at higher cycle numbers. The adsorbed mass estimated using Johannsmanns's model for the cycles 1 and 4 of NC layer in NC-PDDA-HMWL system was 1.11 ± 0.13 mg/m2 and 5.44 ± 1.78 mg/m2, respectively. A similar trend was also observed with the NC-PDDA-OL system with an estimated NC mass of 1.15 ± 0.09 and 5.46 ± 1.79 mg/m2 for cycles 1 and 4, respectively. These estimates suggest that the hydrated mass of NC layer deposited in the 4th deposition cycle is 4x greater than that of the 1st deposition cycle. The increase in mass associated with NC adsorption can also be attributed to the water entrained in the porous structure created by the fibers. An increase in the amount of trapped water, with the increase in layer thickness has been reported in systems involving MFC multilayers48,59. AFM images additionally show the lignin adsorbed along the PDDA coated fibrils after the lignin deposition step, as seen in the images after 3 cycles (Figures 6a and b).

Figure 4. a) AFM Amplitude images of PDDA on mica (5 x 5 μm); b) MWL on PDDA (2.5 x 2.5 μm), and c) OL on PDDA (2.5 x 2.5 μm).

Figure 5. Height images of NC on PDDA after the 1st (a) and 3rd (b) deposition cycle (2.5 x 2.5 μm).

Figure 6. a) Amplitude and b) height images of HMWL after the 4th deposition cycle (2.5 x 2.5 μm). The images show lignin particles deposited on the NC fibrils from the 3rd deposition cycle.
Free-standing LBL Films
Free-standing LBL films were created after 250 deposition cycles with each cycle incorporating two PDDA, one lignin, and one nanocellulose layer. The films were isolated after the cellulose acetate substrate was dissolved in acetone (Figure 7). Initial observation of the film was that it was translucent and bendable. These two properties are rarely associated with lignin-based composites that contain a significant lignin loading. Film samples were dipped in liquid nitrogen for 2 min and applying a bending force with a second set of forceps ruptured the samples. SEM of the cryo-fractured cross-sections of LBL films displays a lamellar structure (Figures 8a and b). The thickness of both NC-PDDA-HMWL and NC-PDDA-OL were found to be approximately 4.3 μm, which implies an average thickness of approximately 17 nm per deposition cycle. The SEM data indicates a significantly higher thickness compared to QCM-D estimates. However, the QCM-D measurements were carried out for only 4 deposition cycles (reaching the limits of the instrument due to the viscoelastic surface). From the QCM-D results, it was noted that the layer build-up was not linear for the four deposition cycles studied. Therefore the data suggests it requires more than 4 deposition cycles for the thickness increase per cycle to plateau.
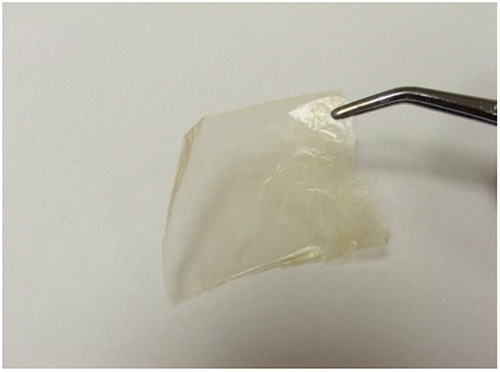
Figure 7. A free-standing film of NC-PDDA-HMWL obtained after 250 deposition cycles. The free-standing films were obtained after dissolving the cellulose acetate substrate in acetone.

Figure 8. SEM images showing lamellar structure of the cross-sections of cryo-fractured LBL films after 250 deposition cycles. The free standing films of a) NC-PDDA-HMWL and b) NC-PPDA-OL were obtained after the cellulose acetate substrate was dissolved in acetone. Please click here to view a larger version of this figure.
Fabrication of Nanocellulose
For nanocellulose fabrication the successful oxidation of the pulp fiber is necessary for facile fibrillation. Oxidation is controlled by available sodium hypochlorite, which should be slowly added at known quantities based on the amount of cellulose. One reason for limited oxidation arises from the storage of the sodium hypochlorite solution for extended periods. This reduced oxidation efficiency can be noted during the reaction; the pulp slurry should turn a pale-yellowish color, part way through the reaction during successful oxidation. If this does not occur, the carboxylic acid content of the fiber is usually below levels that enable easy fibrillation.
The fibrillation of oxidized fiber with carboxylic acid content above 1.0 mmol/g of cellulose can occur by a number of different mechanical treatment methods yielding similar results in nanocellulose particle size. Ultrasonication with a high power sonication horn for short time periods or homogenization with a microfluidic cell are alternatives to blending the oxidized fiber. The former provides a route to prepare single batches of 200 ml of NFC suspensions or less, while the latter provides a route to prepare liters of nanocellulose suspensions. Past experiments using atomic force microscopy have shown that these fibrils have lengths of 530 ± 330 nm and a thickness of 1.4 ± 0.7 nm 60.
QCM-D Viscoelastic Modeling
The mass and thickness of the adsorbed polymer layers can be determined by the Sauerbrey relationship. However, the method is only valid if the deposited layer is rigid, and the whole system performs as a composite resonator. This limitation can be checked by monitoring the frequency dependence of the overtones (ΔF/n). Figure 9 shows that with the increase in the number of layers, the overtones move farther apart, which suggests that as the thickness increases, the response of the films tend to be viscoelastic and less rigid48. For a thicker or viscoelastic film, the nature of propagation of the shear acoustic wave into and through the film affects the estimation of the coupled mass61. Therefore, in such cases, ΔF is not directly proportional to Δm. Also, it is critical to understand that the mass estimated with QCM-D may include coupled water due to hydration and viscous drag. The amount of coupled water varies depending on the nature of the adsorbed film, but can typically range between 1.5-4x the molar mass of the adsorbed material61.
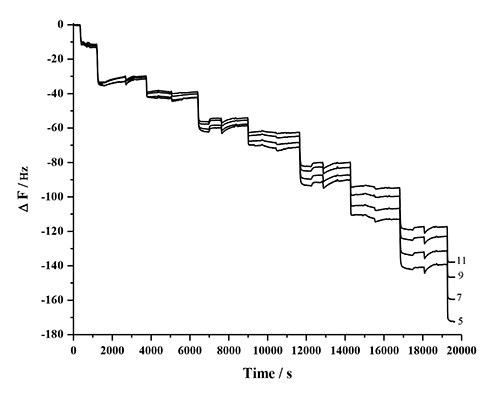
Figure 9. Frequency response of the odd harmonics 5 to 11 as a result of the LBL adsorption of PDDA, HMWL, and NC, showing the frequency dependence of the harmonics as the number of layers increase.
The Johannsmann Model can be used to account for the limitation of the Sauerbrey model. This alternative model determines the true sensed mass of a viscoelastic layer, has been used to study the adsorption characteristics of different systems like polyelectrolyte complexes on polystyrene substrates62, proteins on gold substrate63, and microfibrillated cellulose on polyelectrolytes48,49 . Figure 10 compares the areal mass estimated with Johannsmann model after four deposition cycles for NC-PDDA-HMWL and NC-PDDA-OL systems. The values of the mass adsorbed per cycle and the mass of NC and lignin per cycle are given in Table 1 and 2, respectively. From the comparison, it is seen that the total mass adsorbed after four adsorption cycles is similar for both NC-PDDA-HMWL and NC-PDDA-OL (29.38 ± 2.57 and 31.78 ± 2.44 mg/m2, respectively). From the four adsorption cycles studied, the mass of lignin adsorbed in the first two cycles was seen to differ between the two systems. The mass of HMWL adsorbed in adsorption cycles 1 and 2 were almost twice that of OL (Table 2). However, the mass of the two different lignins adsorbed in cycles 3 and 4 were similar. The mass of NC adsorbed in both systems was similar except for the cycle 2, where the mass of NC adsorbed in NC-PDDA-OL was slightly higher than NC-PDDA-HMWL. This difference caused the beginning of cycle 3 to have the same amount of total mass for the two systems. These results suggest that after the initial cycles there is little difference in assembly for the type of lignin in the composite films. This data suggests that LbL films of model plant walls can be created from lignins of different biological origins and/or isolation protocols. Currently there are not any other methods that can make model stand-alone cell wall materials with select lignins of a particular structure.
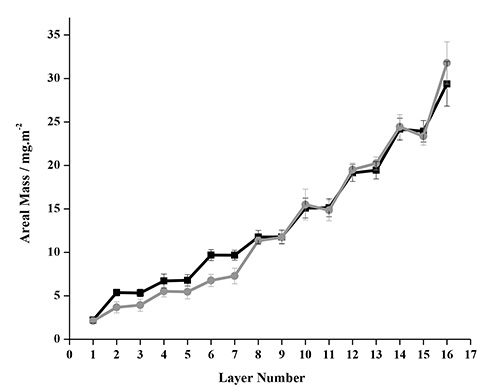
Figure 10. Areal mass estimated with Johannsmann's model for NC-PDDA-HMWL (■) and NC-PDDA-OL (●) after 4 deposition cycles.
| Cycle # | Mass per cycle (mg/m2) | Cumulative mass (mg/m2) | ||
| NC-PDDA-HMWL | NC-PDDA-OL | NC-PDDA-HMWL | NC-PDDA-OL | |
| 1 | 6.72 ± 0.80 | 5.51 ± 0.63 | 6.72 ± 0.79 | 5.51 ± 0.63 |
| 2 | 5.03 ± 0.22 | 5.82 ± 0.50 | 11.76 ± 0.77 | 11.33 ± 0.45 |
| 3 | 7.37 ± 0.37 | 7.52 ± 0.66 | 19.14 ± 0.98 | 19.52 ± 0.73 |
| 4 | 10.23 ± 1.97 | 12.92 ± 1.93 | 29.38 ± 2.57 | 31.78 ± 2.44 |
Table 1. Areal mass estimated from QCM-D data using Johannsmann’s model for 4 deposition cycles.
| Cycle # | Lignin (mg/m2) | Nanocellulose (mg/m2) | ||
| NC-PDDA-HMWL | NC-PDDA-OL | NC-PDDA-HMWL | NC-PDDA-OL | |
| 1 | 3.16 ± 0.26 | 1.56 ± 0.57 | 1.11 ± 0.13 | 1.15 ± 0.09 |
| 2 | 2.91 ± 0.32 | 1.30 ± 0.13 | 2.08 ± 0.36 | 3.18 ± 0.66 |
| 3 | 3.31 ± 0.39 | 3.77 ± 0.14 | 4.00 ± 0.38 | 3.22 ± 1.51 |
| 4 | 4.72 ± 0.64 | 4.22 ± 1.34 | 5.44 ± 1.78 | 5.46 ± 1.79 |
Table 2. Areal mass estimate of lignin and NC using Johannsmann’s model for 4 deposition cycles.
A comparison of the modeled data after four deposition cycles of NC-PDDA-HMWL and NC-PDDA-OL (Figure 11 and Table 3) reveals similar trends observed with Johannsmann’s model. The first trend is the similarity of the final thicknesses between the films with two lignin types for a given film density boundary. The final thicknesses after 4 deposition cycles for NC-PDDA-HMWL and NC-PDDA-OL with an assumed density of 1,000 kg/m2 were 31.5 ± 3.5 and 30.4 ± 5.1 nm, respectively (Figure 12). The final thicknesses for the same, with an assumed density of 1,400 kg/m2, were 23.4 ± 2.8 and 22.1 ± 3.1 nm, respectively. The second trend is revealed at the beginning of the third cycle where the change in thickness is the same for the two lignins, as found for the mass estimate. The thickness of the PDDA layer was not estimated because of negligible or negative changes in the thickness after the adsorption of PDDA. However, it was observed that significant sequential layer build-up was not possible without the adsorption of PDDA following the adsorption of NC or lignin (data not shown). This result indicates that the adsorption of the linking layer is a critical step in the adsorption sequence.
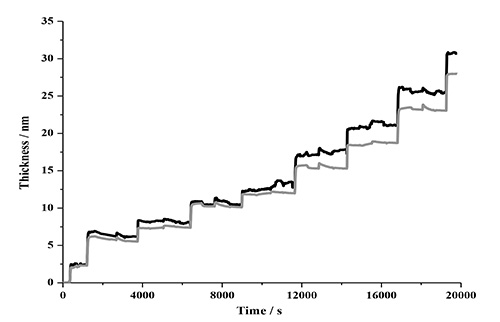
Figure 11. Thickness estimated with Voigt model (black) compared to the thickness estimated with Sauerbrey equation (7th harmonic; grey). The density of the film was assumed to be 1,000 kg/m2.
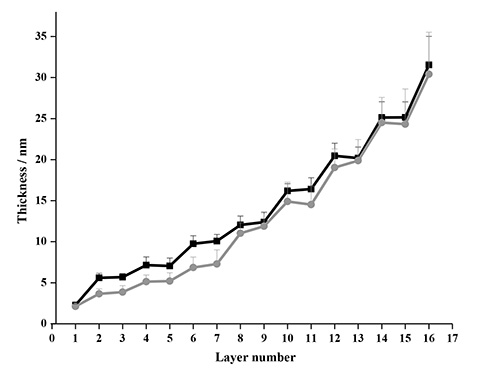
Figure 12. Comparison of thickness estimated with Voigt model for NC-PDDA-HMWL (■) and NC-PDDA-OL (●) after 4 deposition cycles with an assumed density of 1,000 kg/m2.
The thickness of the nanocellulose and lignin films estimated using the viscoelastic model was compared to ellipsometry thickness (dry state) in Table 3. The ellipsometry thickness values from the first deposition cycle gave a close estimate to the Voigt model thickness, with an assumed density of 1,000 kg/m2. The ellipsometry thickness value of the first cycle is almost 2-3x larger than each of the cycles 2-4. This phenomenon is related to the differences in PDDA deposition in the first cycle relative to the other cycles. From the QCM-D experiments, the initial PDDA layer on gold is found to be ~2 nm thick. However, there was negligible or negative change in mass/thickness when PDDA was introduced over NC or lignin. A similar response was seen in a previous study involving LBL adsorption of kraft lignin and PDDA, where a linear buildup of film thickness was observed even though there was negligible PDDA adsorption56. The lower ellipsometry thickness values of the 2nd through 4th deposition cycles compared to the 1st cycle can be attributed to a change in the conformation of the PDDA layer when deposited on gold. However, the relatively small adsorption of PDDA in cycles 2-4 is sufficient to continue the LBL assembly process. There is significant difference between the ellipsometry and QCM-D thickness in the 3rd and 4th cycles, as the estimated Voigt thickness (based on a density of 1,000 kg/m2) was found to be twice that of the ellipsometry thickness for both NC-PDDA-HMWL and NC-PDDA-OL systems. This result relative to the QCM-D data further implicates an increasing viscoelastic nature of the films as the layer build-up proceeds. It is suggested that the topmost layers are more porous compared to the lower layers holding additional trapped water. Moreover, NC used in this study is decorated with carboxyl groups at the C6 position (carboxyl content of 1.0 mmol/g of cellulose), which makes the fibers more hydrophilic. Anionic groups decorating the NC leads to the layer being hydrated and viscous, hence the deviation from the Sauerbrey relation; the relationship is applicable only for thin and elastic films.
| Cycle # | NC-PDDA-HMWL (nm) | NC-PDDA-OL (nm) | ||||
| Voigt | Voigt | Ellipsometry | Voigt | Voigt | Ellipsometry | |
| (1000 kg/m2) | (1400 kg/m2) | (1,000 kg/m2) | (1,400 kg/m2) | |||
| 1 | 7.2 ± 1.0 | 5.0 ± 0.4 | 7.5 ± 0.3 | 5.1 ± 1.0 | 4.0 ± 0.5 | 6.1 ± 0.1 |
| 2 | 12.0 ± 1.1 | 8.8 ± 1.0 | 10.4 ± 0.6 | 11.0 ± 1.3 | 8.0 ± 0.7 | 8.1 ± 0.3 |
| 3 | 20.5 ± 1.5 | 14.4 ± 1.1 | 12.0 ± 0.3 | 19.0 ± 2.3 | 13.2 ± 1.3 | 11.7 ± 0.1 |
| 4 | 31.5 ± 3.5 | 23.4 ± 2.8 | 14.5 ± 0.3 | 30.4 ± 5.1 | 22.1 ± 3.1 | 13.8 ± 0.5 |
Table 3. Cumulative thickness after each adsorption cycle estimated by Voigt model, with assumed densities of 1,000 and 1,400 kg/m3, and thickness estimated by ellipsometry for 4 deposition cycles.
To investigate the validity of the dry film thickness estimated by ellipsometry, an AFM scratch test was performed on both NC-PDDA-HMWL and NC-PDDA-OL after four deposition cycles on a Si wafer. The height profile from the scratch test gave an average thickness of 15.1 ± 0.9 nm and 17.3 ± 3.0 nm respectively for NC-PDDA-HMWL and NC-PDDA-OL, respectively. These values are similar in magnitude to those measured by ellipsometry (Table 3). Ellipsometry resulted in the smallest measurements (optical, dry), followed by AFM (height profile, dry), and QCM-D (sensed mass, estimated from hydrated state).
Lignin Differences
The two types of lignins used in this study were organosolv lignin (OL; Sigma Aldrich, Inc) and hardwood milled wood lignin previously isolated in our laboratories and recently characterized for this current study (HMWL)64. GPC analysis of acetylated samples of HMWL and OL showed an Mn of 5,300 and 1,300 g/mol respectively. The fraction of aromatic:aliphatic acetate hydrogen determined from the 1H NMR analysis of the acetylated lignin samples were found to be 1.16:1, and 0.26:1 respectively for OL and HMWL. Thus, OL was found to have a significantly higher phenolic content, which would account for a greater number of ionizable phenolic groups at an elevated pH. The total acid number of the two lignin determined by conductometric titrations was 0.41 ± 0.02 and 0.34 ± 0.03 mmol/g respectively for OL and HMWL. The acid number determined by conductometric titration represents the contribution from both the phenolic and the carboxylic content present in lignin. Hence the slightly higher charged lignin, which has a lower molecular weight, forms a marginally smaller thickness in the initial frequency change. A difference in deposition is usually noteworthy for polyelectrolyte adsorption onto charged surfaces as segment charge and MW change65. In the first two deposition cycles, organosolv lignin has an areal mass half of the value for the milled-wood lignin. This trend is also observed with the ellipsometry measurements, as thickness of NC-PDDA-OL in the first and second cycle is lower than NC-PDDA-HMWL. However, this study shows that there is minimal change in the third and fourth cycles, as well as when the process is repeated 250x. The great number of cycles will magnify small differences in adsorption. The data suggests that lignin with disparate structure does not greatly impact the fabrication of free-standing films. Hence, either technical lignins, available from biomass conversion into paper, fuels, and chemicals, or model lignins carefully isolated can be used to form free-standing films with nanocellulose. This fact is significant where lignins of different origins can be carefully selected to make model cell wall surfaces.
Future work should integrate hydroxyl rich linker layers (synthetics such as polyvinyl alcohol or biobased like hemicelluloses) to replace or augment the PDDA linker layer used in the current study to derive a structured nanocomposite film that more closely represents the wood cell wall composite. The integration of cellulose microfibrils and lignin across 17 nm is within range of the structures of native cell walls and provides a new model material to serve as an artificial wood cell wall.
