A Simplified Method for Ultra-Low Density, Long-Term Primary Hippocampal Neuron Culture
Summary
Low density cultures of primary hippocampal neurons usually require glia feeder layer to supply neurotrophic factors and sustain longevity. We describe here a simplified method to culture ultra-low density neurons on glass coverslips in the presence of a high density neuronal feeder layer, which facilitates investigation of specific neuronal-autonomous mechanisms.
Abstract
Culturing primary hippocampal neurons in vitro facilitates mechanistic interrogation of many aspects of neuronal development. Dissociated embryonic hippocampal neurons can often grow successfully on glass coverslips at high density under serum-free conditions, but low density cultures typically require a supply of trophic factors by co-culturing them with a glia feeder layer, preparation of which can be time-consuming and laborious. In addition, the presence of glia may confound interpretation of results and preclude studies on neuron-specific mechanisms. Here, a simplified method is presented for ultra-low density (~2,000 neurons/cm2), long-term (>3 months) primary hippocampal neuron culture that is under serum free conditions and without glia cell support. Low density neurons are grown on poly-D-lysine coated coverslips, and flipped on high density neurons grown in a 24-well plate. Instead of using paraffin dots to create a space between the two neuronal layers, the experimenters can simply etch the plastic bottom of the well, on which the high density neurons reside, to create a microspace conducive to low density neuron growth. The co-culture can be easily maintained for >3 months without significant loss of low density neurons, thus facilitating the morphological and physiological study of these neurons. To illustrate this successful culture condition, data are provided to show profuse synapse formation in low density cells after prolonged culture. This co-culture system also facilitates the survival of sparse individual neurons grown in islands of poly-D-lysine substrates and thus the formation of autaptic connections.
Introduction
Growing hippocampal neurons under in vitro conditions enables observation and experimental manipulation of these neurons that are otherwise not possible in vivo. This experimental approach is widely used to reveal neuronal mechanisms of growth, polarity, neurite specification, trafficking and subcellular localization of proteins, synapse formation and functional maturation1. These in vitro cultured hippocampal neurons, when harvested from late embryonic stages, are relatively pure (>90%) glutamatergic cells of pyramidal morphology2. Because neurons were grown in a 2-D surface under in vitro conditions, this method allows easy observation, such as live imaging or immunocytochemistry (ICC) staining through a single focal plane3; or manipulations, such as drug treatment and transfections3-6. When grown at high density, neurons tend to have high rates of survival because of higher concentrations of secreted growth factors in addition to alimentary support from the growth media, and also because of neurite contact-dependent mechanisms7. However, low density hippocampal neurons are desirable for morphological studies, where an individual neuron can be imaged in its entirety or stained for ICC analysis. Low density neurons are hard to maintain in culture due to lack of paracrine support and thus often require trophic support from a glial (typically cortical astrocyte) feeder layer, which has to be prepared prior to neuron culture2. When co-cultured with a glial cell feeder layer, low density neurons are grown on coverslips, and then flipped on top of the glia layer so that the low density neurons and glia are facing each other. A small confined space between glia and neurons is created by placing paraffin wax dots on the corners of the coverslips, therefore creating a 'sandwich' layout2,8,9. The low density neurons will grow within the confined space between glia and the coverslip, which creates a permissive microenvironment with concentrated factors secreted by neurons and glia. This approach yields low-density, fully developed neurons that are spaced reasonably apart, therefore facilitating ICC labeling or live imaging studies.
An apparent drawback of neuron-glia co-culture, aside from being time-consuming and laborious, is that it prevents study of neuron-specific, or cell-autonomous, mechanisms. Although this system is far less complex than in vivo neural tissue, glia impact on neuron development through secreted, not-yet-fully-defined factors can confound the experiments10. Therefore, in experiments that require the investigation of neuron specific mechanisms, defined culture conditions that remove serum and glial support layer are necessary. A previous study has succeeded in culturing low concentrations of neurons (~9,000 cells/cm2) using a three dimensional hydrogel matrix11. Since a relatively pure neuronal population can be cultured at high density under serum free conditions without glial support, we hypothesize that ultra-low density of hippocampal neurons can be grown in serum free defined culture medium by co-culturing them with high density neurons, in a way that is analogous to the conventionally adopted neuron-glia co-culture. Indeed, high density hippocampal neuron cultures in a 'sandwich' configuration have been recently used to support a small number of specialized magnocellular endocrine neurons12.
Therefore, co-culture with high density neurons may allow low density neurons to receive trophic factors support that is sufficient to enable long term survival. This protocol of culturing ultra-low density neurons was thus formulated and validated. The protocol can be implemented within one single experiment, by preparing high density (~250,000 cells/ml) dissociated hippocampal neurons first, and then making a dilution to yield a density ~10,000 neurons/mL (~3,000 neurons/coverslip, or ~2,000 neurons/cm2), which is much lower than most reported low density cultures2,3,9,11,13. This culture condition is loosely referred to as 'ultra-low density' culture and used with 'low-density' inter-changeably. The high density neurons are plated on the poly-D-lysine coated 24-well plates; while the low density neurons are seeded on poly-D-lysine coated 12-mm glass coverslips that are placed inside another 24-well plate. The coverslips with adhering low density neurons are flipped on top of the high density neurons 2 hr later after the neurons are descended and attached to the coverslips. In addition, instead of using paraffin wax dots to elevate the coverslips above the high density neuron layer, an 18 G syringe needle was used to etch the bottom of the 24 well plates with two parallel stripes. The resulting displaced, adjoining plastics provide an elevated support for the glass coverslips. This space is consistently measured at 150-200 μm, which allows sufficient oxygen and culture medium exchange while providing a microenvironment with concentrated trophic factors. Under this condition, the low density neurons grow extensively, and can survive beyond three months in culture. When these neurons are transfected with GFP plasmid after three weeks in culture, the dendrites are profusely studded with dendritic spines. As a proof of principle, data are presented to show that this co-culture system supports ultra-low density cultures of hippocampal neurons seeded on poly-D-Lysine 'micro-islands', where neurons form autaptic connections that may facilitate investigation of cell-autonomous, network-independent mechanisms.
Protocol
All experimental procedures involving mice were approved by the Institutional Animal Care and Use Committee of the University of Arizona, and conformed to NIH guidelines.
1. Tissue Source for Hippocampal Neuron Culture
- To generate prenatal mouse pups for hippocampal neuron culture, use time-pregnant mice (C57Bl6/J) that are bred in house17. The day with vaginal plug detection is designated as E0.5. The planned harvest time for culture is E16.5-E17.5.
Note: This protocol describes cultures of two 24-well plates of two high density neurons co-culturing with low-density neurons (48 coverslips total). For more plates, materials and reagents can be scaled correspondingly.
2. 24-well Plates Preparation for High Density Neuron Cultures
Note: Perform the following steps (2-3) the day before the planned harvest of embryos.
- Bring two 24 well plates into the cell culture hood, open the individual packaging.
- Connect an 18 G syringe needle to a 1 ml plastic single-use syringe.
- Hold the syringe, and aim the needle tip at ~1 mm left to the center of the bottom of the well. Apply moderate force (~250 g) to etch the bottom of the well (~1 mm long). A groove will be formed and the plastic materials displaced will form an elevated support above the flat bottom well surface. Etch another ~1 mm-long parallel groove at ~1 mm to the right of the center of the bottom in a similar way.
- Repeat this step for all the wells of the two 24-well plates. The two 24-well plates used for high density culture are now ready for coating with poly-D-lysine (see step 3.8 below).
3. Coverslips Preparation for Low Density Neuron Cultures
- Use 12-mm diameter #1 round glass.
- Place 50 coverslips inside a 100 mm diameter sterile plastic petri dish, and submerge coverslips in 20 ml 70% ethanol solution. Place this petri dish on a rotating platform with moderate (70 rpm) speed rotation for an hour. Remove 70% ethanol and then replace with a fresh batch of 70% ethanol. Rotate and wash for another hour.
- Discard 70% ethanol cleaning solution. Add sterile water (filtered through 0.2 µm filter unit). Rinse the coverslips rigorously by rotating the petri dish by hand. Rinse three times, each time replacing with fresh sterile water.
- Place the petri dish with coverslips inside a sterile cell culture hood with laminar flow. Aspirate the rinsing water through the vacuum line, and add 10 ml filtered sterile water to immerse the coverslips. The coverslips should be sterile and the surface should be clean enough for poly-D-lysine coating.
- Open two 24-well plates from their individual packing, and place them in the hood.
- Turn on the vacuum line in the hood. Connect a 200 µl pipette tip to the vacuum line, and use a scissor to cut the tip to create a larger opening.
- Use a fine-tip #5 tweezer to pick up coverslips from the petri dish, one at a time, and use the vacuum line to completely dry the coverslip. Then place one coverslip into each of the wells of the 24-well plate. The 50 cleaned coverslips should be enough to fill each of the wells of the two 24-well plates.
- Dilute the poly-D-lysine stock solution (1 mg/ml) into working solution (0.1 mg/ml) with borate buffer (pH 8.5). For the four 24-well plates (including two high density plates with etched bottom), prepare 30 ml of total working solution.
- Add 300 µl 0.1 mg/ml poly-D-lysine work solution to each well with glass coverslips. Make sure the coverslips are completely immersed in solution. If not, use the #5 tweezer tip to press down coverslips against the bottom of the well, and use the tip to guide poly-D-lysine solution to cover all the surface of the coverslips. Visually inspect all the coverslips to make sure no air was trapped between the plate and coverslips.
- Add 300 µl 0.1 mg/ml poly-D-lysine work solution to each well of the high density culture plate with etched bottom. Rotate by hand to spread the solution to cover the entire bottom of the well.
- Return all the four plates with poly-D-lysine to the culture incubator, and incubate O/N.
4. Washing and Pre-conditioning Plates for Culture
Note: The following steps (4-9) are carried out on the day of tissue harvest.
- After incubation/coating of coverslips and 24-well plates O/N, bring all four plates (two with donor coverslips, two acceptor high density plates with etched plastic bottom) into the culture hood. Use the vacuum line to remove the poly-D-lysine solution.
- Immediately following the removal of the poly-D-lysine solution, add ~1 ml sterile water to each well using a plastic transfer pipette. After all the wells are filled with water, let stand for 10 min. Remove water by vacuum, then add 300 µl Wash medium in each well to cover the bottom of the well (with or without glass coverslips). Do not let the poly-D-lysine coating dry out.
- Return all four plates to the cell incubator. The plates are ready to use once the dispersed hippocampal neurons are prepared.
5. Preparation of Complete Culture Medium, and Trypsin Solution for Enzymatic Digestion
- Prepare 60 ml Complete Culture medium by mixing the ingredients (see Materials). Use a 50 ml syringe connected with a 0.2 µm-pore size syringe filter, filter the Complete Culture medium into two 50 ml conical tubes. Each tube contains 30 ml Complete Culture medium.
- In a separate 50 ml conical tube, add ~30 ml Wash medium (see Materials) and warm it up to 37 °C. This will be used to wash the tissue after enzymatic digestion.
- Prepare the enzyme digestion medium. In a 15 ml conical tube, add 4.5 ml Wash medium and 0.5 ml 2.5% trypsin solution, and 50 µl 100X DNase I.
- Place the Complete Culture medium, Wash medium, and enzyme digestion medium in a 37 °C water bath.
6. Preparation of Surgical Tools
- Sterilize all the surgical tools in a sterilization pouch: two small scissors, a pair of forceps and two #5 tweezers.
7. Removal of Brains from E16.5-E17.5 Mouse Embryos, and Dissection of Hippocampi
- Prepare two 10-cm petri dishes filled with ice-cold, sterile Hank's balanced salt solution (HBSS).
- Euthanize the mouse dam with an intraperitoneal injection of sodium phenobarbital (100 mg/kg in a volume of ~ 0.5 ml). After injection, once the dam loses response to toe pinch, sacrifice with cervical dislocation.
- Spray the dam's abdomen area with 70% ethanol, and make a 2-cm long incision along the midline of abdomen to expose the uterus.
- Remove the embryos and place individual ones in a 10-cm diameter petri dish with cold dissecting HBSS buffer.
- Hold the embryo by the neck using a forceps, and use a small scissor to make the first cut right in the midline below the cerebellum level and section across the brain tissue. Do not decapitate the embryo.
- Insert the right side tip of the small scissor from caudal region that is cut open, all the way through the rostral part of the embryonic brain (do not cross midline), and make a cut on the left side at the level of skull base.
- Insert the left side scissor tip again to make a cut on the right side. Leave a narrow band of uncut skull bone and skin tissue at the rostral end, so that the whole brain can be flipped aside for easy extraction.
- Use the tip of scissor to scoop out the embryonic brain into the HBSS solution. Inspect the brain under a dissecting microscope. The brain should be intact with no gross cut-off regions.
- Repeat these steps to collect all the embryo brains.
- Collect all the intact brains in a new petri dish containing 20 ml cold HBSS buffer. Place the petri dish under a dissection microscope.
- View the brain from ventral side. While holding down the brain with forceps at the caudal end, insert a sharp #5 tweezer diagonally to make a cut between the middle rostral point and the caudal-temporal region. Repeat this for the other hemisphere. After cutting, separate two hemispheres with intact hippocampus from the rest of brain stem tissues.
- Repeat this for each of the brains.
- Pool all the dissected hemispheres, and remove the meninges using two pairs of #5 tweezers.
- Identify the developing hippocampus as a curved structure that is folded inside the temporal lobe. Alternate the two pairs of tweezers to separate the hippocampus from adjoining cortical tissues. Collect and pool all the hippocampi tissue (see Figure 1).
8. Enzymatic Digestion, Separate into Single Neurons
- Using a plastic transfer pipette, transfer all the dissected hippocampi into the enzymatic digestion solution containing 5 ml Wash medium with 0.25% trypsin + 1X DNase I in a 15 ml conical tube.
- Place the tube inside a water bath and incubate at 37 °C for 25 min, with intermittent gentle inverting of the tube once in every 5 min.
- After 25 min, carefully remove the enzyme solution using a hand held plastic pipette by aspiration. Add warm Wash medium to 10 ml and gently wash the tissue by slowly inverting the tube 5 times. Remove the Wash medium, and add 10 ml Wash medium again for an additional wash.
- Remove the Wash medium, and add 3 ml of fresh warm Wash medium again. Shake the tube by hand rigorously for 15 times to dislodge the digested neurons from the tissue. The medium should become turbid once the cells are separated from the tissue into the Wash medium. Collect the cell suspension in a new 15 ml conical tube while leaving the remaining tissue in the tube.
- Add another 2 ml Wash medium to the tissue, and gently separate the remaining tissue by triturating through a plastic pipette for ~15 times. Let sit for 5 min. Pool the cell suspension into the new 15 ml conical tube that contains collected 'shake-off' cells.
- Count the density of neurons with a hemocytometer. Typically, 3,000-5,000 cells per microliter can be obtained depending on the number of embryos dissected. An average number of 2.5 x 107 neurons are estimated if assuming six embryos are dissected.
- Calculate the volume needed of this cell suspension to yield a density of 250,000 neurons/ml when added to the 30 ml Complete Culture medium. Add this volume to one of the two conical tubes containing 30 ml Complete Culture medium to yield the high density neuron plating medium.
- Transfer 1.2 ml of this high density neuron solution to another 30 ml Complete Culture medium to yield a concentration of ~10,000 neurons/ml. Use this dilution to seed the low-density coverslips.
9. Plating of Neurons and Long Term Co-culture
- Bring the two 24-well plates with etched bottoms for high density neurons into the cell culture hood. Remove the 300 µl precondition medium by vacuum. Plate 0.6 ml high density cell suspensions at 250,000 neurons/ml.
- Bring the other two 24-well plates with coverslips into the culture hood, and remove the 300 µl precondition medium by vacuum. Plate 0.6 ml low density cell suspensions at 10,000 neurons/ml on the coverslips inside the two 24-well plates.
- Return all four 24-well plates to the incubator. Let sit for 2 hr.
- Flip the low density coverslips with adhering neurons to the high density culture. Make sure the neurons are facing each other (i.e., not separated by the glass coverslip). Then return the two plates with co-culture to the incubator.
10. Co-culture Sustaining
- Feed co-cultures by adding 300 µl fresh Feed medium (see Materials) at day in vitro (DIV) 5. There is no need to remove 50% of original Complete Culture medium, as reported by many other studies. The feeding medium also contains 15 µM cytosine arabinoside (Ara-C, to yield a final concentration of 5 µM) to minimize the chance of glia contamination and proliferation.
- Following this initial feeding, add 300 µl fresh Feed medium without Ara-C to the well every week. Should the culture be kept for more than one month, replace half of the medium with fresh Feed medium every week beyond one month time point (with the first medium half-replacement at DIV33). Morphologically, both high density and low density neurons look healthy up to three months (the longest time observed by the experimenters).
11. Illustration of an Experimental Manipulation-low Density Neuron Transfection
Note: When transfection in low density neurons is desired, a simple calcium phosphate protocol can be adopted. We have found that low density cultures have better transfection efficiency with calcium phosphate protocol before DIV12, but they can be transfected with much lower efficiency at DIV21 or older, during which time dendritic spines are prominent. The feasibility of transfection of the cultured low density neuron is illustrated by transfecting neurons with a pEGFP-C3 plasmid (see below).
- At DIV21, remove 600 µl conditioned medium from each well of the co-culture, and transfer it to a new 24-well plate. Then add 600 µl fresh Feed medium to the co-culture to bring it back to the original volume.
- Transfer the coverslips into the new 24-well plate with 600 µl conditioned medium. Make sure the low density neurons are facing upwards. Place the high density plates and the new plates with coverslips back in the incubator.
- Prepare two 1.5 ml sterile tubes. In one tube, add 600 µl 2X HEPES buffered saline (HBS) solution. Calculate the volume of 12 µg pEGFP-C3 DNA based on plasmid concentration. In the other tube, add 74 µl CaCl2 (2 M stock) and the volume of water that after adding plasmid will make 600 µl. Mix well by pipetting and then add plasmid DNA. Slowly mix well. Let both tubes sit for 5 min at RT.
- While hand-holding the 2X HBS tube and moderately agitating the solution on a mixer, add all of the 600 µl the DNA-CaCl2 solution drop by drop to the 600 µl 2X HBS tube. It is critical that the precipitate formed is in the shape of 'fine sand' granules. Mixing too strong and too fast is not desirable, since it will more likely produce large 'snow flake'-like precipitates, which has dramatically lower transfection efficiency based on our observations.
- Let the precipitate continue to form in the dark at RT for 30 min.
- Add 100 µl precipitate to each well that contains low density neuron on the coverslips, drop by drop and evenly. Return the plate to incubator, and incubate for 2 hr. Assuming most of the CaCl2 is in free form, this results in a concentration of ~17.6 mM Ca2+, which may be toxic to neurons following prolonged incubation.
- Prepare a 50 mL conical tube with ~50 ml Wash medium, warm it up to 37 °C.
- At the end of 2 h, bring the low density neuron with DNA-calcium phosphate precipitates into the hood. Pick up each individual coverslip with a sharp #5 tweezer, and dip it three times in the 50 ml Wash medium to wash it briefly. Then return it to the high density neurons in its original co-culture plates, with low density neurons facing downwards.
- Continue the culture until the low density culture neurons on coverslips are harvested for studies.
Representative Results
The protocol described here enables successful ultra-low density, long-term culture of pure glutamatergic neurons without the need of glia cells serving as a feeder layer. The protocol is diagramed in Figure 1, which involves preparation of high density (on poly-D-lysine coated 24 wells) and low-density neurons (on poly-D-lysine coated glass coverslips) separately, and subsequent co-culture that can be maintained up to three months.
Figures 2A and 2B are illustrations of the high density and low density neurons at 5 days after plating. Low density culture is approximately 30 fold less in density. Both neurons undergo typical developmental stages as outlined by Kaech and Banker (2006). At day 14, both high density and low density neurons show elaborate dendritic structures, as revealed by DIC images (Figures 2C, D, note both panels are from the same field with different focal planes, as shown by the location of etch). When these neurons are stained with MAP2 antibody to reveal the dendrites, no significant differences in the number of dendrites (t13= 1.27, p > 0.05; measured by dendritic tip number) and total dendritic length (t16= 1.41, p > 0.05) per neuron between the high density and low density neurons are found (Figure 2E, F).
Immunocytochemistry labeling is used to reveal functional glutamatergic synapses. We use double staining to label the NMDA receptor subunit NR1 and the AMPA receptor subunit GluR1 (Figure 3A), and functional synapses (co-localized puncta in yellow) can be readily identified and quantified using ImageJ. Low density neurons are also labeled with antibody against dendritic protein marker MAP2, in combination with axonal protein marker p-Tau. This double-labeling allows clear distinction of dendrites and axons (Figure 3B). Low density cultures can also be successfully transfected with DNA plasmids using calcium phosphate methods, although the transfection rate is typically very low at later stages (< 0.2% neurons when transfected at DIV21). Figure 3C illustrates that EGFP transfection can reveal neuronal morphology, and render fine dendritic spine structures visible. Lastly, as a proof of concept, ultra-low density neurons can be grown under conditions that promote autapse formation (Figure 3D). These ultra-low density autaptic neurons grown on poly-D-lysine coated 'micro-islands' can be sustained by the high density feeder neuron to beyond 2 months with this co-culture protocol.
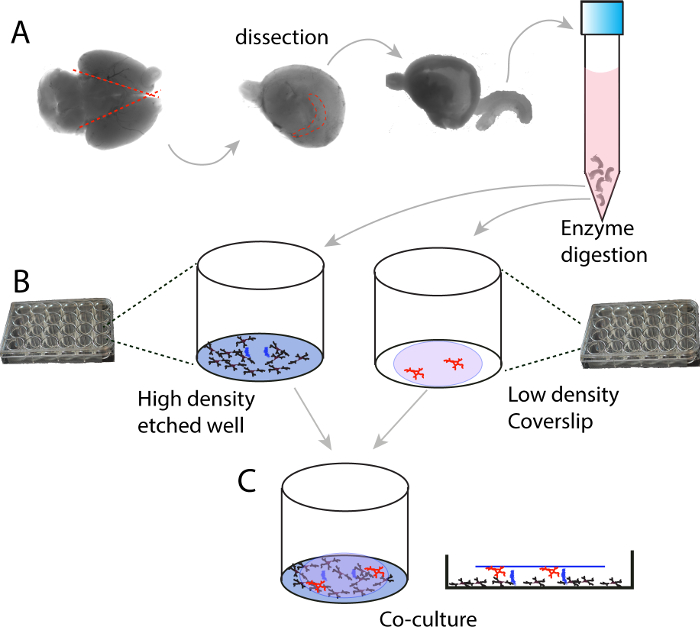
Figure 1. Schematic illustration of the high density and low density co-culture protocol. (A) E16.5-E17.5 mouse embryonic brains are collected, hippocampi are dissected out and digested with trypsin. (B) Digested hippocampi are washed, separated into single neurons, and plated at high density (250,000 cells/ml) on 24 well plates; in the meanwhile, low density (10,000 cells/ml) neurons are plated on coverslips. The wells for high density culture are etched with an 18 G syringe needle to provide an elevated support for the coverslips. (C) Illustration of the co-culture. Please click here to view a larger version of this figure.
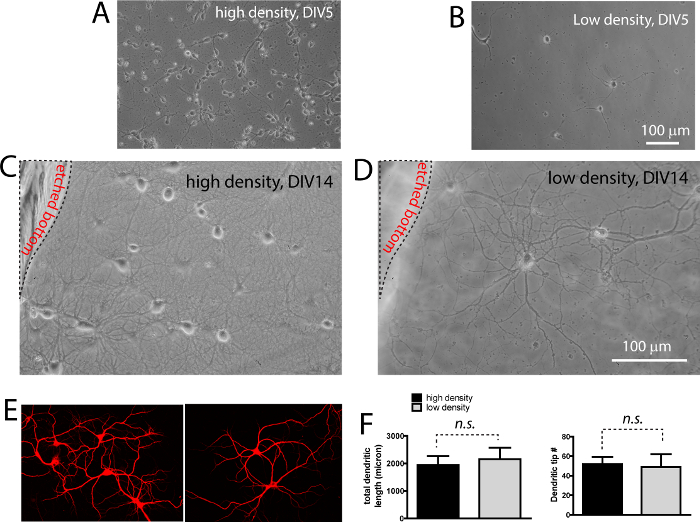
Figure 2. Both high density and low density neurons have comparable morphological development in co-culture. (A, B) Photomicrographs showing plating density of both high density and low density layer. (C, D) DIC images showing extensive neurite growth in both high (C) and low density (D) neurons. The low density neurons on coverslip are resting right above the high density neurons. Note the location of the raised plastic support. (E) Immunocytochemistry labeling of dendritic protein MAP2. (F) Quantification (mean ± s.e.m) of total dendritic length and dendritic tip number per neuron. No significant difference (n.s.) was seen between high density and low density neurons grown in co-culture. Scale bar in (B, D), 100 μm. Please click here to view a larger version of this figure.
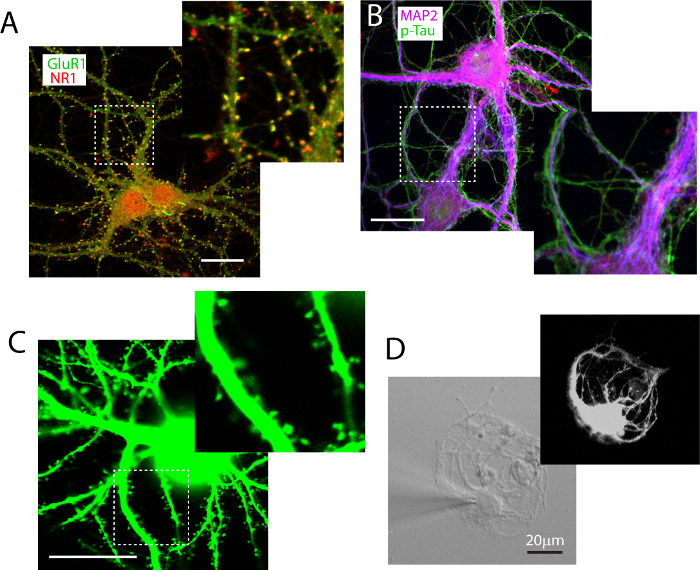
Figure 3. Experimental manipulations of low density neurons grown in co-culture. (A) Immunocytochemistry labeling in low density culture allows identification of functional synapse defined by co-labeling of GluR1 (green) and NR1 (red). (B) Co-labeling of neuronal dendritic protein MAP2 (magenta) and axonal protein p-Tau (green). (C) A low-density hippocampal neuron transfected with pEGFP-C3 plasmid, showing profuse dendritic spines. (D) A low density neuron prepared on poly-D-lysine 'micro-island', and grown on top of the high density neurons. A recording patch electrode fills the neuron with Alexa-555 hydrazide to reveal the morphology of the neuron. Scale bar in A-D, 20 μm. Please click here to view a larger version of this figure.
Discussion
We present a detailed protocol for long-term culture of ultra-low density hippocampal glutamatergic neurons under serum free conditions. At ~2000 neurons/cm2, the density is at least two fold lower than most 'low density' culture preparations with or without glia support reported by the existing literature2,3,11,13,14. In addition to being ultra-low density, this protocol is novel and significant in two more ways. First, no glia feeder layer is needed as the low density neurons obtain trophic factor support from the high density neurons that are within close physical proximity, even though there are no synaptic connections or direct contact between the neurons cultured at different densities. This protocol eliminates the need for preparing glia monolayer ahead of the planned culture experiments, and allows both high density and low density cultures to grow simultaneously. Preparation of the glia monolayer can be time-consuming and laborious, and most literature uses neonatal rat pup cortices2. Therefore, for labs that work with mice only, this requires significant additional efforts. More importantly, in experiments where neuronal specific mechanisms are investigated, the presence of glia in the co-culture can be undesirable, because it prevents ascribing observed effects specifically to neurons, glia or the interaction between the two cell types10. In addition, for glia culture, serum supplement is obligatory2,15, and this may contaminate the glia-neuron co-culture and introduce additional confounding factors. One important feature of our protocol is that the co-culture is under defined medium conditions. We strictly use the Complete Culture medium without serum supplement, and find that if the culture medium exchange schedule is strictly followed, the co-culture can be sustained beyond three months, which should satisfy the majority of experiments. Because there is virtually no glia presence, one limitation of this technique is that caution should be exercised when comparing with the results of literature that use glia-neuron co-cultures.
Another novel aspect of this protocol is that we use a simple method to create an effective micro-space between the high density neurons grown on the bottom of the 24-well plate, and the low density neuron adhered to the glass coverslips sitting right above. Previous studies uses paraffin dots applied to the coated coverslips to raise the coverslips at ~500 μm above the glia feeder layer2,8,9. Preparation of paraffin dots requires the right size and right temperature, which demands a reasonable amount of efforts and practice. In comparison, a simple etching on the well bottom plastics with an 18 G needle provides support for the coverslips as well as good separation between the two layers of neurons. We have used a patch clamp recording rig with digital Z meter to determine the linear space between the two layer of neurons by focusing on the high density and then on the low density layers using a 60X water immersion objective, and found that the space is typically 150-200 μm (179.4 +/-25.3 μm, n = 5). This narrower space (compared with that generated by wax dots) is likely to translate to a faster rise, and higher concentration of neurotrophic factors, therefore providing sufficient support for the low density neuron to grow. Although this narrower space may raise questions about the rate of culture medium exchange and whether the smaller space impedes neurons from obtaining oxygen and nutritional support, our results show that this is not likely a concern. On the contrary, we found that the high density neurons under the coverslips also grow better (higher survival rate, more elaborate dendritic tree and more synaptic puncta by NR1 staining, data not shown) compared with the high density neurons from the same preparation but without the overlaying coverslips. Therefore, this simple protocol is not only fast, simple, but also very effective in sustaining both high density and low density neurons.
Culturing hippocampal neurons in vitro requires coating of growth promoting substrates, such as poly-D-lysine and/or laminin/collagen5,16. Although meticulous selection and preparation of glass coverslips are critical for effective neuronal survival, we have found that with the cover glass we selected, a thorough cleaning with 70% ethanol is sufficient, thus negating the need of harsher treatment (e.g., boiling in acids) of the coverslips. After the coverslips are cleaned in ethanol and dried by vacuum, the glass seems to be hydrophobic due to the original coating. However, successful coating of 0.1 mg/ml poly-D-lysine in borate buffer should render the glass surface hydrophilic, so that when the coverslips are picked up from the washing water, a uniform layer of water that is evenly spreading across the entire glass surface can be observed. We found that this is critically important. Otherwise the neurons will most likely die after plating due to insufficient coating. Although only low density hippocampal neurons are tested in this protocol, it is expected that other neuronal types (e.g., cortical neurons, cerebellar granule neurons, or specialized neural populations) can also be sustained at low density in the presence of a high density neuron feeder layer. Lastly, when this simple protocol is followed, one can expect to harvest healthy ultra-low density neurons. Broad applications such as immunocytochemistry labeling, live imaging, morphological studies and electrophysiology recording can all be conducted on these neurons following standard procedures16,17.
Declarações
The authors have nothing to disclose.
Acknowledgements
This study was supported by an NIH/NIMH grant to S.Q. (R00MH087628).
Materials
| Neurobasal medium | Life Technologies | 21103-049 | Protect from light |
| B27 supplement | Life Technologies | 17504-044 | aliquot, store in 0.6ml size |
| GlutaMAX-I | Life Technologies | 35050-061 | dilute 100X |
| antibiotic-antimycotic (AA) | Life Technologies | 15240-096 | dilute 100X |
| Complete Culture medium | Neurobasal medium with 1X B27, 1X AA, 1X GlutaMAX-I | ||
| Wash medium | same as 'Neurobasal medium' | ||
| Feed medium | Neurobasal with 1X B27 supplement | ||
| DNAse I | Sigma-Aldrich | D5025 | prepare 100X stock at 0.6mg/ml |
| poly-D-lysine | Sigma-Aldrich | P6407 | M.W. 70000-150000 |
| borate buffer | Sigma-Aldrich | B6768 (boric acid); 71997(borax) | 1.24g boric acid & 1.9g borax in 400ml H2O, pH to 8.5 use HCl |
| 12-mm round glass coverslips | Glasswarenfabrik Karl Hecht GmbH | 1001/12 | No. 1 glass, purchase from Carolina Biological Supply |
| proFection transfection kit | Promega | E1200 | see protocol for details |
| 2X HEPES buffered saline (HBS) | Promega | E1200 | see protocol for details |
| Syringe filter | Pall Corporation | 4192 | 0.2um pore size |
| Endofree plasmid prep kit | Qiagen | 12362 | for preparation of transfection grade plasmid DNA |
| anti-MAP2 antibody | Millipore | MAB3418 | mouse antibody, clone AP20 |
| anti-p-Tau antibody | Millipore | AB10417 | rabbit polyclonal antibody |
| anti-NR1 antibody | Millipore | MAB1586 | mouse antibody, clone R1JHL |
| anti-GluR1 antibody | Millipore | AB1504 | rabbit polyclonal antibody |
| Hank's balanced salt solution | ThermoFisher | 14025092 | 500ml size |
Referências
- Banker, G. . Cultureing Nerve Cells. , 339-370 (1998).
- Kaech, S., Banker, G. Culturing hippocampal neurons. Nat Protoc. 1, 2406-2415 (2006).
- Petersen, J. D., Kaech, S., Banker, G. Selective microtubule-based transport of dendritic membrane proteins arises in concert with axon specification. J Neurosci. 34, 4135-4147 (2014).
- Seibenhener, M. L., Wooten, M. W. Isolation and culture of hippocampal neurons from prenatal mice. J Vis Exp. , (2012).
- Shi, Y., Ethell, I. M. Integrins control dendritic spine plasticity in hippocampal neurons through NMDA receptor and Ca2+/calmodulin-dependent protein kinase II-mediated actin reorganization. J Neurosci. 26, 1813-1822 (2006).
- Viesselmann, C., Ballweg, J., Lumbard, D., Dent, E. W. Nucleofection and primary culture of embryonic mouse hippocampal and cortical neurons. J Vis Exp. , (2011).
- Biffi, E., Regalia, G., Menegon, A., Ferrigno, G., Pedrocchi, A. The influence of neuronal density and maturation on network activity of hippocampal cell cultures: a methodological study. PLoS One. 8, e83899 (2013).
- Crump, F. T., Dillman, K. S., Craig, A. M. cAMP-dependent protein kinase mediates activity-regulated synaptic targeting of NMDA receptors. J Neurosci. 21, 5079-5088 (2001).
- Leal, G., Afonso, P. M., Duarte, C. B. Neuronal activity induces synaptic delivery of hnRNP A2/B1 by a BDNF-dependent mechanism in cultured hippocampal neurons. PLoS One. 9, e108175 (2014).
- Clarke, L. E., Barres, B. A. Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci. 14, 311-321 (2013).
- Kaneko, A., Sankai, Y. Long-term culture of rat hippocampal neurons at low density in serum-free medium: combination of the sandwich culture technique with the three-dimensional nanofibrous hydrogel PuraMatrix. PLoS One. 9, e102703 (2014).
- Millet, L. J., Bora, A., Sweedler, J. V., Gillette, M. U. Direct cellular peptidomics of supraoptic magnocellular and hippocampal neurons in low-density co-cultures. ACS Chem Neurosci. 1, 36-48 (2010).
- Salama-Cohen, P., Arevalo, M. A., Meier, J., Grantyn, R., Rodriguez-Tebar, A. NGF controls dendrite development in hippocampal neurons by binding to p75NTR and modulating the cellular targets of Notch. Mol Biol Cell. 16, 339-347 (2005).
- Xu, S. Y., Wu, Y. M., Ji, Z., Gao, X. Y., Pan, S. Y. A modified technique for culturing primary fetal rat cortical neurons. J Biomed Biotechnol. 2012, 803930 (2012).
- McCarthy, K. D., de Vellis, J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 85, 890-902 (1980).
- Qiu, S., Weeber, E. J. Reelin signaling facilitates maturation of CA1 glutamatergic synapses. J Neurophysiol. 97, 2312-2321 (2007).
- Qiu, S., Lu, Z., Levitt, P. MET receptor tyrosine kinase controls dendritic complexity, spine morphogenesis, and glutamatergic synapse maturation in the hippocampus. J Neurosci. 34, 16166-16179 (2014).
