Following the preparation of each SLB, septins or the protein of interest may be incubated with the desired support and imaged via TIRFM, confocal microscopy, or SEM. The results shown here use septins recombinantly expressed and purified from E. coli17. Using TIRFM on planar SLBs, it is possible to determine the length of filaments and their flexibility, measure the diffusion coefficients and observe assembly over time28,29. In order to collect the highest quality measurements, it is first necessary to ascertain the quality of bilayers, especially when preparing them for the first time or when changing lipid compositions. A visual inspection of the protein distribution on the bilayers by TIRFM can help identify regions of the bilayer that have been scratched or are malformed. Protein distribution should be homogeneous (Figure 3A), and there should not be holes or gaps in the bilayer (Figure 3B). It is best to avoid membranes with holes, which can form from dust contamination or smudges from slide handling, as this can change the protein distribution in other areas of the bilayer. To visualize the membrane itself, trace rhodamine-PE can be incorporated into SUVs for bilayer formation. In high-quality bilayers, the field will appear even (images not shown), but if liposomes are old or if washes are not stringent enough, unburst and tubulated liposomes may accumulate on the surface (Figure 3C). Additionally, while solid supports will hamper the free diffusion of lipids8, the lipids should not be immobile. FRAP experiments can be used to assess the mobility of lipids on planar bilayers, which may vary by composition and should show recovery rates in the order of seconds30.
Spherical supports can be used to examine protein binding on membranes of defined curvatures either in isolation (one curvature) or with several membrane curvatures in the same well to observe competition between curvatures6,18. Near-TIRFM on beads >1 µm can also be used to measure the number of association events for a given area27. As with planar bilayers, we use rhodamine-PE to look for smooth bilayer deposition (Figure 4A), i.e., no lipid clumps, which indicate multi-lamellarity or unburst liposomes (Figure 4B), and no gaps in the bilayer (Figure 4C). For measuring the total protein adsorption or the protein adsorption over time on curved surfaces, it is necessary to isolate individual beads from each other in order to create a discrete volume for which sum lipid intensity and sum septin intensity can be measured. Thus, the beads should be well-separated rather than clumped together as in Figure 4D. We address troubleshooting options for all of these potential issues in the discussion section.
This SLB assay can also be applied to rod supports, which offer an environment for proteins to sample multiple curvatures on a single surface. Pairing this assay with scanning electron microscopy allows the user to examine curvature preference, alignment, and length distribution of septins18 or other proteins of interest. While similar to the other assays presented here, the rod assay does not allow for careful control of total membrane surface area because of the heterogeneous nature of the substrate that is derived from filter paper. The material properties of the filter paper used here result in rods of different lengths and diameters (Figure 5A); this is useful for exploring protein curvature sensing and organization on curved surfaces (Figure 5B), but because the ratio of protein to the membrane cannot be controlled, the rods are of limited utility for generating saturation-binding curves or measuring parameters such as binding constants.
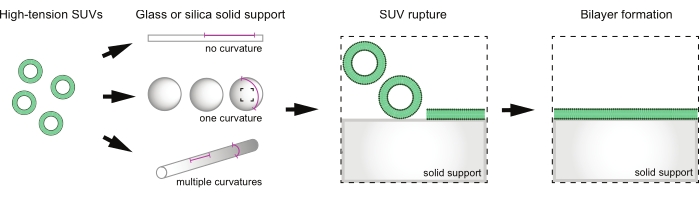
Figure 1: Overview of supported lipid bilayer formation on supports with various curvatures. SUVs are incubated with solid supports of different geometries in order to change the curvatures available for sampling on a given membrane. SUVs adsorb onto the solid support surface and rupture to create lipid bilayers. Please click here to view a larger version of this figure.
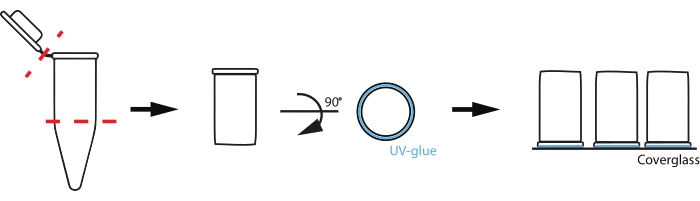
Figure 2: Schematic of reaction chamber set up. To prepare custom chambers, a 0.2 mL PCR tube is cut where the tube begins to taper and at the cap (red dashed lines). The uncut rim of the cut tube is then coated with a thin layer of UV-activated glue (blue) and placed glue-down on a coverslip. Please click here to view a larger version of this figure.
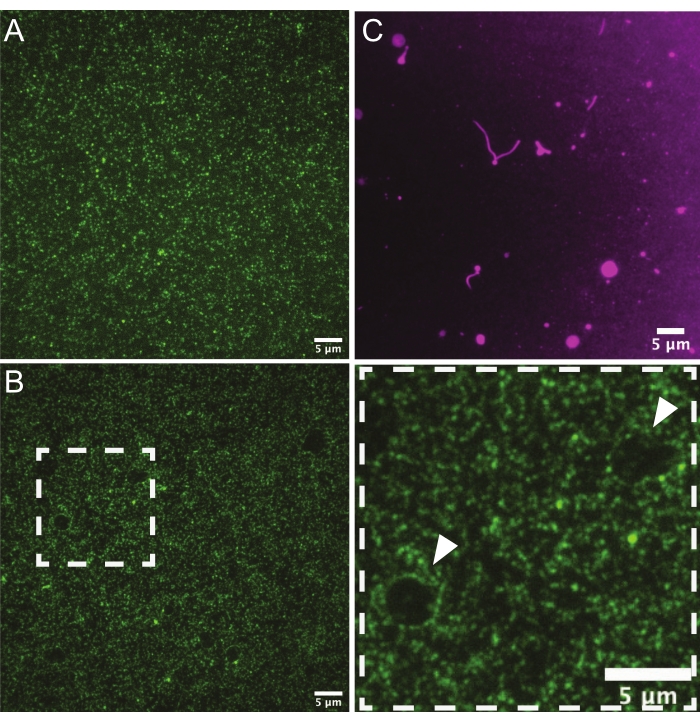
Figure 3: Representative TIRF micrographs of planar bilayers. (A) A representative image of a high-quality lipid bilayer (75% DOPC, 25% Soy PI)with non-polymerizable septins bound (green). Protein is not clustering in any specific regions, and there are no liposomes attached to the membrane and no holes. (B) This bilayer was made with poorly cleaned glass coverslips and exhibits what appear to be "holes" (white arrows) in the membrane where there are fewer septins. Septins can be seen crowded at the edges of these defects in the bilayer (denoted by the white arrows in the zoomed region on the right). (C) A representative image of a low-quality bilayer using trace rhodamine-PE. Unburst liposomes and tubulated lipids are visible on the surface. Please click here to view a larger version of this figure.
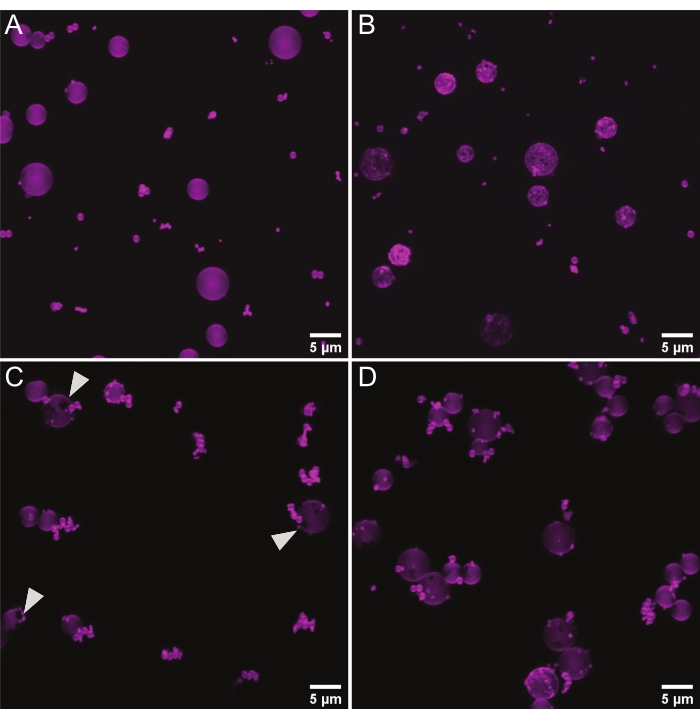
Figure 4: Representative micrographs of lipid-coated microspheres. Representative images from competition assays where lipid-coated beads (75% DOPC, 25% Soy PI, 0.1% Rhodamine PE)with diameters of 0.3 µm, 0.5 µm, 1 µm, 3 µm, and 5 µm were mixed. All images are maximum Z projections. (A) Representative image of a high-quality mixture of lipid-coated microspheres. The spherical supports are evenly coated by the membrane, and there are few bead clusters. (B) Representative image of uneven membrane coating, likely caused by insufficient washing of excess lipids. (C) Representative image of beads with gaps in membrane coverage (white arrows) due to improper handling. (D) Representative image of densely clustered beads, likely caused by insufficient mixing throughout the procedure, especially when combining the different beads together. Please click here to view a larger version of this figure.

Figure 5: Representative electron micrographs of lipid and septin-coated rods. (A) SEM image showing the distribution of lipid-coated (75% DOPC, 25% Soy PI, 0.1% Rhodamine PE) rod lengths and diameters. (B) The edge of an isolated membrane-coated rod with septin filaments aligned along the axis of positive curvature. Please click here to view a larger version of this figure.
