Quantification of Bacterial Histidine Kinase Autophosphorylation Using a Nitrocellulose Binding Assay
Summary
We report and demonstrate an optimized nitrocellulose binding assay that can be used to quantify autophosphorylation of purified bacterial histidine kinases. Our method has several advantages over traditional SDS-PAGE based techniques, providing a valuable alternative for characterizing these important proteins.
Abstract
We demonstrate a useful method for quantifying autophosphorylation of purified bacterial histidine kinases. Histidine kinases are known for their involvement in two-component signal transduction, a ubiquitous system through which bacteria sense and respond to environmental stimuli. Two-component signaling features autophosphorylation of a histidine kinase, followed by phosphotransfer to the receiver domain of a response regulator protein, which ultimately leads to an output response. Autophosphorylation of the histidine kinase is responsive to the presence of a cognate environmental stimulus, thereby giving bacteria a means to detect and respond to changes in the environment. Despite their importance in bacterial biology, histidine kinases remain poorly understood due to the inherent lability of phosphohistidine. Conventional methods for studying these proteins, such as SDS-PAGE autoradiography, have significant shortcomings. We have developed a nitrocellulose binding assay that can be used to characterize histidine kinases. The protocol for this assay is simple and easy to execute. Our method is higher throughput, less time-consuming, and offers a greater dynamic range than SDS-PAGE autoradiography.
Introduction
Adaptive response is crucial for bacterial survival. In order to detect and respond to environmental changes, bacteria use a stimulus-response system known as two-component signaling.1,2 In a typical two-component system, the histidine kinase detects a cognate stimulus, autophosphorylates its conserved histidine residue, then transfers phosphate to a conserved aspartate residue on the receiver domain of a response regulator protein.3 This event triggers a change in the activity of the response regulator, which stimulates a downstream effect.4,5 Thus, bacteria are able to sense and adapt to changes in the local environment. Some two-component signaling systems deviate from this archetype. In some cases, the sensory domain of the histidine kinase is a stand-alone protein, which directly detects the sensory input and modifies kinase activity through a protein-protein interaction.6–8 However, the fundamental process and overall role of the system remains the same. Two-component signaling is a ubiquitous stimulus-response system that is essential for bacterial survival, and histidine kinases play a critical role in the transduction of the signal.9
Despite the importance of histidine kinases to bacterial biology, they remain poorly characterized. This is due to the inherent instability of phosphohistidine, and the lack of a practical method for measuring autophosphorylation. Phosphohistidine is more labile than phosphoserine, phosphothreonine, and phosphotyrosine.10 Thus, techniques that are commonly used to analyze Ser/Thr/Tyr kinases are not applicable for histidine kinases.11 In vitro assays to study histidine kinases have largely been limited to SDS-PAGE autoradiography.12,13 In this method, [γ-32P]-ATP is incubated with the kinase, and phosphorylation of the kinase is analyzed by polyacrylamide gel electrophoresis (PAGE) followed by autoradiography of the gel. This method can be used to monitor kinase autophosphorylation, as well as phosphotransfer from the kinase to a response regulator. However, this method has notable shortcomings. PAGE-based assays are low throughput and time-consuming. Such limitations are not conducive to characterizing a protein and ascertaining its kinetic parameters. An alternative method for studying histidine kinases that was published recently utilizes phosphohistidine antibodies to detect autophosphorylation.14 While this method has the advantage of distinguishing between 1-phosphohistidine and 3-phosphohistidine, depending on the instrumentation used for detection, this method may not offer a large dynamic range or high upper limit of detection. Thus, there is a need for a faster, less laborious, and more sensitive assay that can be used to study these important proteins.
Here, we describe and demonstrate a carefully developed nitrocellulose binding assay that can be used to quantify autophosphorylation of purified bacterial histidine kinases in vitro. This assay is higher throughput and less time-consuming than PAGE-based assays. The method also utilizes Cherenkov radiation for phosphohistidine quantification, which offers a high upper limit of detection and a large dynamic range. The assay can be used to determine kinetic parameters for histidine kinases.
Protocol
Caution: This protocol requires appropriate training in the use and handling of radioactive materials. Please use the requisite personal protective equipment when performing this assay, including beta radiation shielding. Radioactive waste must be handled carefully, as large volumes of waste are generated during the washing phase of the experiment. Ensure that the waste is stored in an upright container such as a large bucket or bottle that will not be easily knocked over or spilled. Once the experiment is concluded, carefully transfer all liquid waste to appropriately marked radioactive waste containers. Handle all materials with care, and keep a Geiger counter nearby to monitor the workspace for contamination.
NOTE: This protocol is a revised version of a previously reported assay from our group.15 Phosphohistidine stability in H3PO4 should be tested for any uncharacterized histidine kinase prior to using this method. The phosphohistidine stability test has been described previously.15 A negative control that does not contain kinase must be included. This is necessary in order to subtract the background signal from each sample, and ensure that [γ-32P]-ATP is sufficiently washed from the membrane.
1. Preparation of Reagents and Materials
- Purify histidine kinases from bacterial culture prior to using this assay.
- Purify a histidine kinase (gene ID 1189383) from Vibrio parahaemolyticus (EB101, ATCC 17802) for the development of this assay. Clone the kinase into expression vector pET-23aHis-TEV using NdeI and XhoI restriction sites, and carry out site-directed mutagenesis to yield the mutant construct Vp1876 D499A.
- Transform the plasmid into E. coli BL21 (DE3) pLysS, and grow cells in TB media at 37 °C. Supplement cultures with ampicillin (100 µg/mL) and chloramphenicol (34 µg/mL), and grow with agitation (250 rpm) to an OD600 of 0.6.
- Induce protein expression with IPTG to a final concentration of 25 µM, and grow cultures overnight at 16 °C. Harvest cells by centrifugation (5,000 x g), lyse by sonication, and centrifuge to remove cellular debris (18,000 x g).
- Purify the His-tagged kinase using Ni-NTA agarose. Confirm purity by SDS-PAGE. Optimize purification for each protein, and confirm purity prior to using this assay.
- Prepare the 25 mM H3PO4 wash solution. To 1 L of distilled deionized water, add H3PO4 to a final concentration of 25 mM. The pH of this solution is approximately 2.0. Place 25 mM H3PO4 on ice.
- Prepare a 13x stock solution of 325 mM H3PO4, to be used to quench kinase reaction. The pH of this solution is approximately 1.5. Place 325 mM H3PO4 on ice.
NOTE: H3PO4 at a final concentration of 25 mM sufficiently quenches the reaction, and also helps to block radiolabeled phosphate that is not bound to histidine from binding to the nitrocellulose membrane. - Prepare a 4x reaction buffer stock solution containing 160 mM Tris-HCl pH 8.0, 600 mM KCl, 16 mM MgCl2, and 40% glycerol.
- Quantify histidine kinase concentration by established protein concentration methods (Bradford, UV-Vis, etc.).16,17 Prepare a histidine kinase solution with a concentration that is 4 times the desired final concentration. To obtain adequate signal, the final protein concentration in the reaction should ideally be at least 5 µM.
- Prepare a 4x radiolabeled [γ-32P]-ATP solution with the appropriate 4x concentration of unlabeled ATP and labeled:unlabeled ratio for the experiment.
NOTE: The amount of [γ-32P]-ATP that should be included in this mix is dependent on how much [γ-32P]-ATP will ultimately be spotted for each reaction. We have obtained adequate signal with final concentrations ranging between 0.1 – 5 µCi per reaction. It is important that the labeled:unlabeled ATP ratio be kept constant for all reactions in a given experiment. Serial dilutions should be made when multiple ATP concentrations are desired, as this well keep the labeled:unlabeled ATP ratio constant across all concentrations. Final ATP concentrations typically range from 10 µM to at least 1 mM, and each concentration should be assayed in triplicate to obtain reliable kinetic data. - 96- well dot blot apparatus
- Cut an 8 cm x 12 cm piece of nitrocellulose membrane (0.2 µm pore size).
- Position nitrocellulose in a dot blot apparatus, ensuring that the membrane fits such that the apparatus is sealed, and all wells are completely covered by the membrane. If assembled correctly, there should be no air leakage when the vacuum is applied.
- To collect filtrate, connect the apparatus to a secondary side-arm flask. From the valve stem, connect adequate tubing such that the apparatus can be comfortably used without tipping the secondary flask.
- Connect the secondary side-arm flask to aspirator/vacuum source.
- Test that the apparatus is completely sealed by applying vacuum to the apparatus. If the apparatus is correctly assembled, a strong vacuum will be present in all of the wells. All tubing connections can be wrapped in Parafilm or saran wrap to ensure a tight seal.
- Optionally, to test the vacuum, pipet 100 µL of reaction buffer into one well. The vacuum should be strong enough to draw all liquid through the well and onto the membrane.
- Turn off vacuum until all samples are ready to be spotted on the membrane.
2. Reaction Initiation and Quenching
- Mixing of reaction components
- Prior to initiation, prepare all four reaction components as 4x stock solutions: reaction buffer (see section 1.4), histidine kinase (1.5), [γ-32P]-ATP (1.6), and ddH2O.
- Mix equal volumes of reaction buffer, histidine kinase, and ddH2O. Allow these reaction components to equilibrate at room temperature for a short time (10 min).
NOTE: The final reaction volume is dependent on whether the experiment is time-dependent, enzyme-dependent, or substrate-dependent. Time-dependent reactions must be larger, as multiple aliquots are taken from the same reaction and quenched at the desired time point. The reaction volume will depend on the number of time points desired. The assay is optimized for each spot on the nitrocellulose membrane to contain 30 µL of the reaction. Thus, if 10 time points are desired, the reaction volume must be at least 300 µL (a higher volume such as 330 µL would be preferable in the event of any pipetting errors). Enzyme and substrate-dependent experiments require smaller reaction volumes. The reaction volume for these experiments can be as small as 30 µL, as this is the final volume that will be spotted on the nitrocellulose membrane. - To initiate the reaction, add one volume of [γ-32P]-ATP solution to the reaction and mix by pipetting up and down. Track elapsed reaction time with a timer and allow the reaction to proceed for the desired time.
- To quench the reaction, add 1/13 of the total reaction volume of ice-cold 325 mM H3PO4 to the reaction and mix by pipetting up and down. Alternatively, add an aliquot of the reaction to 325 mM H3PO4.
NOTE: The final concentration of H3PO4 should be 25 mM to sufficiently quench the reaction. For example, add 2.5 µL 325 mM H3PO4 to 30 µL reaction, or add 30 µL reaction to 2.5 µL 325 mM H3PO4. The final pH of the reaction is approximately 4.0. This concentration of H3PO4 has been tested and confirmed to quench the kinase reaction in this buffer without degrading phosphohistidine.- For example, mix 7.5 µL 4x reaction buffer, 7.5 µL 4x histidine kinase, and 7.5 µL ddH2O. Initiate reaction with 7.5 µL [γ-32P]-ATP. Quench the reaction with 2.5 µL 325 mM H3PO4.
- Immediately place quenched reactions on ice until all reactions have been terminated.
NOTE: To maximize efficiency, it is advisable to initiate one reaction every 15 or 20 s until all reactions are initiated, and once the desired reaction time has passed, quench one reaction every 15 or 20 s until all are quenched. For those who are using the assay for the first time, a longer interval may be more manageable.
3. Spotting of Quenched Reactions on Nitrocellulose
- Start vacuum on 96-well dot blot apparatus
- Place the pre-assembled 96-well dot blot apparatus (section 1.7) into a secondary container. This container should be large enough for the apparatus to be easily disassembled in, as the apparatus and membrane will be radioactive after use.
- Apply vacuum to the 96-well dot blot apparatus.
- Once the apparatus is under vacuum, carefully pipet the quenched reaction directly onto the nitrocellulose membrane in each well. Repeat until all reactions are loaded onto the membrane. Since the binding capacity of nitrocellulose (75 – 110 µg/cm2) is greater than the amount of histidine kinase that is spotted, it can be assumed that nearly all of the kinase binds to the membrane when loaded correctly.
NOTE: Depending on the apparatus used, care must be taken not to puncture the nitrocellulose. Observe that all of the reaction has made contact with the nitrocellulose. It is easy for some or all of the reaction to stick to the walls of the well, and not reach the nitrocellulose. - Wash the wells with 100 µL of ice-cold 25 mM H3PO4. This will allow any reaction volume that may have been trapped in the well to reach the membrane, ensuring complete loading of all reactions. Allow the entire volume to flow through the membrane.
- Apparatus disassembly and nitrocellulose membrane transfer
- Carefully disassemble the apparatus before shutting off the vacuum. Be advised that both the apparatus and nitrocellulose membrane are radioactive at this time. Do not remove any apparatus components from the secondary container at this time.
- With forceps, carefully transfer the nitrocellulose membrane from the apparatus to a container of approximately 200 mL 25 mM ice-cold H3PO4. Place a lid on this container, as the wash will be radioactive. At this time, the vacuum may be shut off.
4. Nitrocellulose Processing
- Nitrocellulose washing
- Place the container with the washing membrane on a rocker. Allow the membrane to gently rock for 20 min.
- After 20 min, carefully decant the used wash solution into a large bucket to be used for temporary waste storage. Add 200 mL of ice-cold 25 mM H3PO4 to the membrane and repeat.
NOTE: At least three 20 min washes are necessary to remove background [γ-32P]-ATP signal from the membrane. After the third wash, test the used wash solution for radiation with a Geiger counter. Continue washing the membrane as described above until no signal is present in the wash solution.
- Nitrocellulose drying
- After the membrane is sufficiently washed, allow the membrane to briefly air dry. This typically takes 5 min or less.
5. Exposure to Storage Phosphor Screen
Note: This section is optional. Exposing the membrane to a phosphor screen is beneficial in that it allows for visualization of the intensity of radiolabeled kinase in each spot on the membrane. The relative intensity of these spots is directly proportional to the amount of phosphorylated histidine kinase in each spot. The intensity can be quantified with image processing software, and these results can be compared to those generated from section 7. Furthermore, this step allows for quality control. Abnormalities seen in this scan might explain irregular results obtained from scintillation counting.
- Phosphor screen preparation
- Prior to exposing the membrane to the storage phosphor screen, expose the phosphor screen to white light for at least 5 min. This ensures that any residual image that may be held by the screen is erased.
- Make sure the screen is clean and dry. If necessary, gently clean the screen with a phosphor screen cleaning solution approved by the screen manufacturer, and wipe dry.
- Nitrocellulose membrane preparation
- Carefully wrap the dry nitrocellulose membrane in a thin plastic wrap to prevent residual moisture from damaging the phosphor screen. Ensure that there are no wrinkles or bubbles.
- Exposure to phosphor screen
- Place the wrapped nitrocellulose membrane in the storage phosphor screen cassette, place the screen face-down onto the membrane, and close the cassette. Do not open the cassette or move the membrane until it is time to scan the phosphor screen, as membrane shifting during exposure will cause a double or smeared image to be captured.
- Expose for at least 4 h, or as long as the phosphor screen manufacturer suggests.
- Once exposure is completed, scan the phosphor screen and capture the image with a phosphor scanner.
6. Preparing Nitrocellulose Membrane for Scintillation Counting
- Ponceau S staining and destaining
- Briefly immerse the nitrocellulose membrane in Ponceau S staining solution (0.1% Ponceau S (w/v) in 5% acetic acid) for 1 – 2 min. Wet the entire membrane with the stain prior to destaining.
- Decant excess Ponceau S from the membrane.
- Rinse the membrane with ddH2O until only the spots from the histidine kinase are stained. Note that this procedure will only work if all spots contain detectable amounts of protein.
NOTE: This step is necessary to confirm that the kinase is bound to the nitrocellulose, and that protein loading is even throughout all spots. It also allows the spotted kinase to be easily cut out from the membrane.
- Cutting out spots
- Using scissors, cut out each spot on the nitrocellulose membrane. It is not necessary to cut perfectly along the edge of the stained spot; if the membrane was washed sufficiently, background due to varying size of cut out spots will be negligible. It is only important to cut out the entire spot for each reaction.
- Using forceps, carefully transfer each spot into scintillation vials. No scintillation cocktail is necessary as the 32P is readily detectable by Cherenkov radiation.
NOTE: Alternatively, the spot can be cut out using a sharpened cork borer. Remove each spot from the nitrocellulose with the cork borer, and push it into a scintillation vial with a pin. With this method, the membrane does not need to be touched, further minimizing radiation exposure.
- Determining CPM/pmol of [γ-32P]-ATP solution
- Spot several dilutions of [γ-32P]-ATP solution (section 1.5) onto 1 cm x 1 cm squares of nitrocellulose. Briefly air dry.
- Using forceps, carefully transfer each square into scintillation vials. No scintillation cocktail is necessary. These samples will be used to generate a standard curve to determine the CPM/pmol of the ATP solution.
7. Scintillation Counting
- Load all scintillation vials into scintillation counter cassettes. Load cassettes into scintillation counter.
- Execute scintillation counting program to measure CPM for each vial. These data, along with the CPM/pmol of the radiolabeled ATP mix (from section 6.3), can be used to calculate the amount of phosphorylated histidine kinase present in each spot, and thus the rate of autophosphorylation.18,19
Representative Results
A representative data set was generated featuring an image captured with a phosphor imager (Figure 1), Ponceau S stain of the nitrocellulose membrane (Figure 2), and scintillation counting data (Figure 3). Figure 3A shows the enzyme kinetic constants in a Lineweaver-Burk plot. These results were obtained using a purified histidine kinase from the gram-negative species Vibrio parahaemolyticus (gene ID 1189383). The protocol used to purify this kinase is described in section 1.1. The final kinase concentration in the reaction was 7.5 µM. The final ATP concentrations ranged from 0 µM to 1.28 mM. The [γ-32P]-ATP solution was 171.97 CPM/pmol ATP. These data can all be generated in a single day using the procedure we have described.
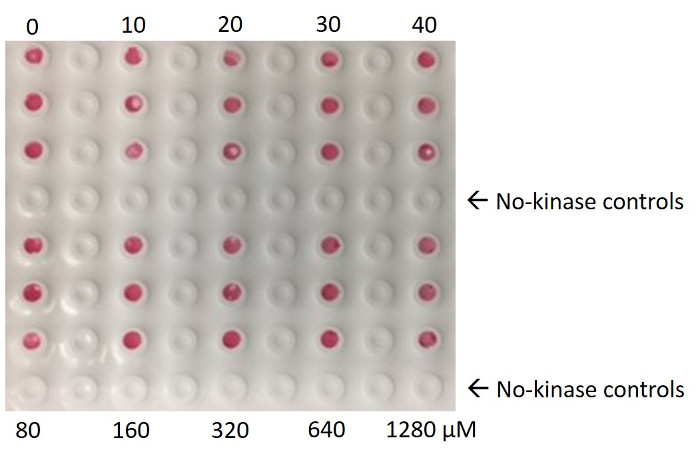
Figure 1: Ponceau S staining of a nitrocellulose membrane. Ponceau stain showing the kinase bound to the nitrocellulose membrane. The membrane was spotted with reactions containing various concentrations of ATP, but constant kinase concentration. No protein is detected in the no-kinase control spots (4th and 8th row). Ponceau S staining solution was composed of 0.1% Ponceau S (w/v) in 5% acetic acid. Please click here to view a larger version of this figure.

Figure 2: Fluorography results. Scanned image of a phosphor screen that had been exposed to a nitrocellulose membrane. The membrane was spotted with reactions containing constant kinase concentration, and various concentrations of ATP. Each concentration was assayed in triplicate. At each concentration, a no kinase control was included (4th and 8th row) to demonstrate that radiolabeled ATP is completely removed from the membrane during the washing step. Please click here to view a larger version of this figure.
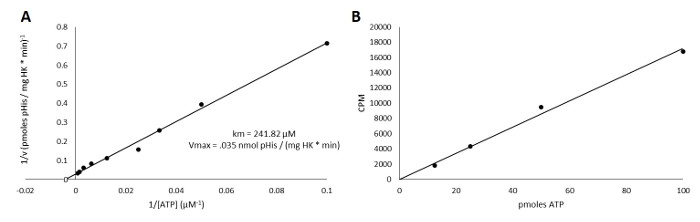
Figure 3: Scintillation counting data. A) Double-reciprocal plot of the autophosphorylation of histidine kinase. The substrate-dependence curve was generated from scintillation counting. Phosphohistidine formation was calculated using the slope of Figure 3B (CPM/pmol ATP), which was also generated with scintillation spectrometry. B) Standard curve of the CPM/pmol of ATP. The slope is used to calculate the amount of phosphohistidine (pmol) present in each sample. Please click here to view a larger version of this figure.
Discussion
The nitrocellulose binding assay we have described has many advantages over previously used methods to characterize histidine kinases. In comparison to traditional SDS/PAGE-based autoradiography, our method is higher throughput and less time-consuming. The nitrocellulose membrane is easier to handle than SDS gels, and does not need to be fixed. Ponceau staining the nitrocellulose allows for the protein spots to be visualized. This provides an easy way to cut out each spot for scintillation counting, and determine that protein loading is consistent across all spots. Scintillation counting of each spot provides accurate results that can be easily converted to reaction velocity using the slope of the standard curve shown in Figure 3B.
Although exposing the nitrocellulose membrane to a storage phosphor screen adds time to the total duration of the experiment, this step offers insight into the success of the blotting. Any discrepancies that are noticed in the scintillation counting data can potentially be explained by the complementary phosphor image. If, for instance, the membrane was not sufficiently washed, the phosphor image will reveal this information. Fluorography of the membrane is recommended to anyone who plans to use this assay, as the results from this step can be a valuable piece of supplemental information.
Another significant advantage of this assay is the ability to generate data from scintillation counting. Cutting out each individual spot for scintillation counting is preferable to use of image processing software to quantify band intensity, as is typically done for PAGE-based assays. The upper limit and dynamic range for detection are far higher with scintillation counting. Generating a standard curve to determine the CPM/pmol ATP make it easy to calculate the rate of autophosphorylation from the raw data. Using this method, we are able to accurately detect picomoles of phosphorylated product.
Despite the many advantages of using our method, it is not without its limitations. Our method does not distinguish between 1-phosphohistidine and 3-phosphohistidine. Although phosphohistidine antibodies can be used to distinguish between these phosphorylated products, our method still has the advantage of utilizing Cherenkov radiation to quantify phosphorylation. Cherenkov radiation provides a large dynamic range and high upper limit of detection. Depending on the method of detection, using antibodies to quantify phosphohistidine formation may suffer from a lesser upper limit and dynamic range. Our method is also limited to being used with purified proteins, and can't be used to detect phosphorylated kinases in vivo. Our method requires radiolabeled substrate, which may deter labs that are not equipped for radioactivity. Ultimately, this method is mostly suitable for labs that are already equipped to handle radioactivity, and are interested in a higher throughput and less time consuming alternative to frequently used SDS-PAGE techniques to study histidine kinases. Those who are studying Ser/Thr/Tyr kinases may also find this method to be useful, despite the relative abundance of alternative methods for studying these proteins.
Characterization of histidine kinases has long been elusive despite advances in methods to quantify other types of kinases. With this assay, we may begin to characterize these biologically relevant yet poorly understood proteins. This advancement in methodology for quantifying histidine kinase autophosphorylation will ultimately improve our understanding of two-component signaling in bacteria. This assay will be a valuable tool as we explore the effect of cognate stimuli and protein-protein interactions on histidine kinase activity in various two-component signaling pathways.
Declarações
The authors have nothing to disclose.
Acknowledgements
This work was supported by the Department of Education through the Graduate Assistance in Areas of National Need program (P200A100044).
Materials
| Phosphoric acid | VWR | AAAA18067-AP | For quenching reactions and washing nitrocellulose |
| Tris base | RPI | T60040-5000.0 | Tris-HCl pH 8.0, for kinase reaction buffer |
| Potassium chloride | RPI | P41000-2500.0 | For kinase reaction buffer |
| Magnesium chloride | RPI | M24000-500.0 | For kinase reaction buffer |
| Glycerol | RPI | G22020-4000.0 | For kinase reaction buffer |
| 5'-ATP | Promega | E6011 | Kinase substrate |
| [γ-32P]-5'-ATP | Perkin Elmer | NEG002Z250UC | 6000 Ci/mmol |
| 96-well dot blot apparatus | Bio-rad | 1706545 | For spotting reactions |
| Nitrocellulose | Whatman | 32-10401396-PK | For spotting reactions |
| Ponceau S | Sigma aldrich | P3504-50G | For staining nitrocellulose |
Referências
- Stock, A. M., Robinson, V. L., Goudreau, P. N. Two-component signal transduction. Annu. Rev. Biochem. 69, 183-215 (2000).
- Jung, K., Fried, L., Behr, S., Heermann, R. Histidine kinases and response regulators in networks. Curr. Opin. Microbiol. 15 (2), 118-124 (2012).
- Parkinson, J. S., Kofoid, E. C. Communication modules in bacterial signaling proteins. Annu. Rev. Genet. 26, 71-112 (1992).
- Laub, M. T., Biondi, E. G., Skerker, J. M. Phosphotransfer profiling: systematic mapping of two-component signal transduction pathways and phosphorelays. Methods Enzymol. 423, 531-548 (2007).
- Ronson, C. W., Nixon, B. T., Ausubel, F. M. Conserved domains in bacterial regulatory proteins that respond to environmental stimuli. Cell. 49 (5), 579-581 (1987).
- Szurmant, H., Bu, L., Brooks, C. L., Hoch, J. A. An essential sensor histidine kinase controlled by transmembrane helix interactions with its auxiliary proteins. Proc. Natl. Acad. Sci. USA. 105 (15), 5891-5896 (2008).
- Price, M. S., Chao, L. Y., Marletta, M. A. Shewanella oneidensis MR-1 H-NOX regulation of a histidine kinase by nitric oxide. Bioquímica. 46 (48), 13677-13683 (2007).
- Zhulin, I. B. The superfamily of chemotaxis transducers: from physiology to genomics and back. Adv. Microb. Physiol. 45, 157-198 (2001).
- Robinson, V. L., Buckler, D. R., Stock, A. M. A tale of two components: a novel kinase and a regulatory switch. Nature Struct. Biol. 7 (8), 626-633 (2000).
- Attwood, P. V., Piggott, M. J., Zu, X. L., Besant, P. G. Focus on phosphohistidine. Amino acids. 32 (1), 145-156 (2007).
- Kee, J. -. M., Muir, T. W. Chasing phosphohistidine, an elusive sibling in the phosphoamino acid family. ACS Chem. Biol. 7 (1), 44-51 (2012).
- Tawa, P., Stewart, R. C. Kinetics of CheA Autophosphorylation and Dephosphorylation Reactions. Bioquímica. 33 (25), 7917-7924 (1994).
- Marina, A., Mott, C., Auyzenberg, A., Hendrickson, W. A., Waldburger, C. D. Structural and mutational analysis of the PhoQ histidine kinase catalytic domain. Insight into the reaction mechanism. J. Biol. Chem. 276 (44), 41182-41190 (2001).
- Fuhs, S. R., et al. Monoclonal 1- and 3-Phosphohistidine Antibodies: New Tools to Study Histidine Phosphorylation. Cell. 162 (1), 198-210 (2015).
- Ueno, T. B., Johnson, R. A., Boon, E. M. Optimized assay for the quantification of histidine kinase autophosphorylation. Biochem. Biophys. Res. Commun. 465 (3), 331-337 (2015).
- Bradford, M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 7 (72), 248-254 (1976).
- Stoscheck, C. M. Quantitation of protein. Methods Enzymol. 182, 50-68 (1990).
- Funt, B. L., Hetherington, A. Scintillation counting of beta activity on filter paper. Science. 131 (3413), 1608-1609 (1960).
- Bem, E. M., Bem, H., Reimchüssel, W. Determination of phosphorus-32 and calcium-45 in biological samples by Čerenkov and liquid scintillation counting. Int. J. Appl. Radiat. Isot. 31 (6), 371-374 (1980).
