1. Preparing Culture Solutions and Media (not in video)
- Prepare 1 liter of Complete Hank’s balanced salt solution (HBSS) containing 1x HBSS, 2.5 mM Hepes (pH7.4), 30 mM D-glucose, 1 mM CaCl2, 1 mM MgSO4 and 4 mM NaHCO3. Add double distilled water (ddH2O). Filter sterilize with a 0.2-μm filter and store at 4 °C.
- Prepare slice culture medium using 35 mL of Basal Medium Eagle media, 12.9 mL of Complete HBSS, 20 mM D-glucose (1.35 mL of 1 M solution), 1 mM Glutamax (0.25 mL of a 200 mM solution), 0.5 ml of Penicillin-streptomycin 100x stock. Filter sterilize with a 0.2-μm filter, then add heat-inactivated horse serum to a final concentration of 5%.
- Prepare Laminin working solution by making a 1 mg/mL Laminin stock solution with sterile distilled deionized water. Prepare 100 μL aliquots in 0.5 mL eppendorf tubes and freeze at -80 °C.
- Prepare Poly-L-lysine working solution by adding 5 mL of sterile H2O to 5 mg of poly-L-lysine to make a 1 mg/mL stock solution. Prepare 1 mL aliquots and freeze at -20 °C.
- Prepare coating solution by diluting 1 mL of poly-L-lysine and 100 μL of laminin to a final volume of 12 mL with sterile water. Make this solution fresh each time.
2. Preparing Organotypic Slice Inserts (not in video)
- Prepare two six-well plates with one culture insert per well using sterile forceps. Add 2 mL of sterile ddH2O underneath the culture inserts.
- Add 1 mL of the coating solution on top of the membrane taking care not to puncture the membrane. Incubate overnight in a humidified incubator at 37 °C and 5% CO2.
- Remove coating media and wash membrane with sterile H20 three times. Let inserts dry before use. Add 1.8 mL of slice culture medium and place in 37 °C incubator. Wrap the unused plates with Parafilm and store at 4 °C for up to 4 weeks.
3. Preparing for Electroporation (not in video)
- Prepare RNAi constructs for electroporation. Double strand RNA hairpin inserts were cloned into a pSilencer vector. The plasmid contains: 1) a U6 promoter that drives the double strand RNA generation; and 2) a GFP expression cassette driven by the CMV promoter8,9. This plasmid has been previously described by Konishi and colleagues9 and others7,8. Plasmids are purified using a Qiagen maxi-prep kit, and used at a concentration of 1 mg/mL.
- In order to visualize DNA while injecting it, prepare a 0.5% fast green dye solution and use it at 1:20 with the DNA to be injected (usually 20 μL of DNA with 1 μL of fast green). Left over DNA-fast green dye mixture can be stored at -20 °C for up to a week.
- Clean dissection area, dissecting tools and vibratome vessel with 70% ethanol. Chill complete HBSS so that it is ice cold. Chill vibratome vessel by packing ice around it inside the vibratome with some water for rapid cooling. Prepare 3% low melting point agarose using complete HBSS. Microwave for 1 min. Avoid over boiling. Keep in a 42 °C waterbath until use.
- Electroporation parameters are set as follows. For an E15 embryo use 35 V, 5 pulses, 100 ms length, 900 ms interval between pulses. For older animals, to ensure electroporation, use higher voltage up to 50 V or increase the number of pulses up to 8 pulses. To prevent damaging the tissue in younger animals, use either fewer pulses or up to 2 pulses and lower voltage up to 25 V. These parameters may be varied and determined empirically depending on the age of the animal.
4. Dissection and Electroporation (in video)
- After euthanizing a pregnant female, dissect embryos out into ice cold complete HBSS. Keep each embryo in their individual placental sacs.
- Dissect embryo out and cut off the head after the first vertebra. Keep in ice cold complete HBSS.
- For injection, place the head on a piece of Parafilm on top of a petri dish. Using custom made Hamilton syringe (see Table I) inject about 6 to 8 μL of DNA:fast green dye mix through the third ventricle in order to fill in both lateral ventricles in the cortical vesicles. Alternatively, injection can be done directly into each lateral ventricle.
- For the ex vivo electroporation, use BTX-tweezer platinum electrodes. Place the positive electrode towards the side of the cortex you want to electroporate i.e. top of the head for dorsal cortex.
- After electroporation, incubate the heads on ice for at least 5 min before dissecting.
- Dissect brains in ice cold HBSSby making a small incision on the side of the head and peeling the skin off the sides of the head. Next, with fine forceps gently peel away the pia from the brain. Remove the intact brain from the skull, taking care not to damage the cortex.
5. Embedding and Sectioning of Electroporated Cortices (in video)
- Transfer 3% low melting point agarose into a large mold placed on ice. The bottom of the mold will start to solidify faster which will prevent brains from sinking down to the bottom of the mold. Gently, transfer brains with fine forceps one by one after removing excess buffer with a Kimwipe or filter paper. Use a 10 μL pipet tip to swirl the brains inside the mold to ensure maximum interface between the agarose and brain tissue.
- Orient the brains to ensure all brains are in the same orientation and at about the same level in the agarose. Let the agarose solidify for about 5 min. Use bonding adhesive (crazy glue) to attach the agarose blocks so that the olfactory bulbs are standing up. Once the blocks are attached, immediately add ice cold HBSS and trim the agarose to make sure individual slices are obtained for each brain.
- To slice the blocks, set the vibratome’s velocity to a low speed (about half the maximum) and set the blade vibration frequency at the highest setting. Generate 250-μm thick coronal slices. Retrieve slices using a bent fine spatula and transfer them to tissue wells with a fine brush or forceps.
- In tissue culture hood, transfer slices into coated inserts. Add 500 μL of slice culture media to each insert to make the transfer easy. Up to 5 slices can be placed per insert. Remove excess media from the top of the slices and incubate at 37 °C in humidified incubator.
6. Culture and Analysis of Organotypic Slices (in video)
- In order to maintain healthy slices, fresh media should be added at least every other day underneath the membrane by replacing half of the media each time.
- In order to analyze slices after the desired days in culture, fix slices in the membrane. Wash with 1x phosphate saline buffer (PBS) at 37 °C three times for 10 min each time. Next, fix with 4% Paraformaldehyde (PFA) overnight at 4 °C or for 1 hr at room temperature.
- Slices can be analyzed with different cellular markers or stained with Hoechst alone to visualize electroporated and non-electroporated cells. Permeabilize and block slices for 2 hr at room temperature with 10% goat serum, 0.1% triton in 1x PBS with gentle shaking.
- Stain with Hoechst for 1 hr at room temperature, wash 3 times with 1x PBS 10 min each time with gentle shaking.
- To mount slices cut the membrane with a scalpel, and use a fine forceps to transfer the membrane containing slices to a frosted glass slides in a water chamber. Up to 5 slices can be placed per glass slide. Remove excess water, and add a drop of Flourmount solution to each brain slice. Gently place a coverslip on top of the brain slices and remove any air bubbles. Analyze slices using a confocal microscope.
7. Alternative Paraffin Embedding of Organotypic Slices (not in video)
- Organotypic slices can also be embedded for paraffin for a finer morphological analysis, to this end the membrane containing the organotypic slices can be fixed in 4% PFA as described above.
- The fixed slices are embedded in 1% agarose (pre-warmed to 37 °C), and solidified in ice for 30 min. The agarose blocks can then be post-fixed in 4% PFA at 4 °C for 30 min.
- The agarose block containing the organotypic slice will then be embedded in paraffin and processed for immunofluorescence as previously described10.
8. Representative Results
A diagrammatic representation of the electroporation of murine cortex and culture of organotypic slices is shown in Figure 1. This method is a useful strategy for rapid assessment of the function of genes involved in neuronal development11. Depending on the amount of DNA electroporated and the embryonic stage at electroporation, the transfection efficiency will vary. Slices will start expressing GFP at least 8 hours post-electroporation and cells will undergo the normal sequence of neurogenic events (proliferation, migration, and early neuronal differentiation) in culture. Figure 2 shows an electroporated brain slice that is expressing a control pSilencer- GFP vector and one can observe neuronal progenitors, migrating neurons and differentiated neurons in the slice. Organotypic slices will keep their morphology as long as they are maintained in a good media-air interface on the membranes and can be used up to at least 5 days in culture.
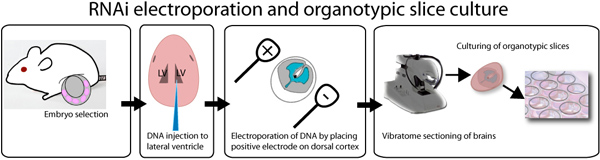
Figure 1. Illustration of ex vivo electroporation and organotypic slice culture assay. E14.5 embryos are dissected out, and individually injected with DNA mixed with fast green dye in order to visualize the injection site. DNA can be injected in both the lateral ventricles as depicted in the illustration or in the third ventricle in order to fill the lateral ventricles. After injection, brains are electroporated with a square wave electroporator, placing the positive electrode on the desired side of the brain. Brains are embedded in 3% low melting point agarose and sectioned using a vibratome. 250 μm brain slices are placed on 0.4 μm inserts and cultured up to a week. GFP can be observed after 8 hours post transfection.
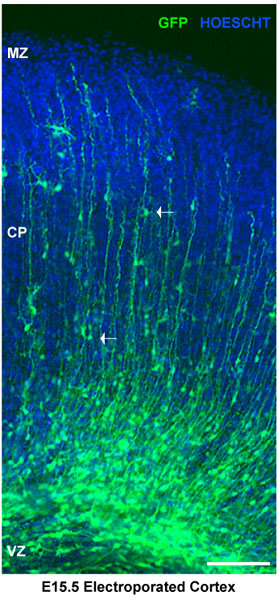
Figure 2. Analysis of electroporated brain slices. Electroporated brain slices were stained for Hoescht. Dorsal cortex shows electroporated neuronal progenitors at the ventricular zone (vz). Neurons in the cortical plate (cp) are delimited by the marginal zone (mz). White arrows show migrating neurons. In this case, brains were injected at E15.5 and electroporated with a pSilencer GFP control vector. The sections represent cortical explants 4 days after electroporation. Scale bar 100 μm.
Troubleshooting:
- Low transfection efficiency: Adjust the concentration of DNA used to at least 1 μg/μL. Always use very clean DNA from a maxi prep if necessary use an endo-free Quiagen kit to purify the DNA.
- Cells transfected in a different brain area than the one desired: Ensure that the electrodes are positioned correctly with the positive electrode position towards the side of the brain to be electroporated.
- Organotypic slices lose morphology: Change media every day and ensure the slices are not floating in media
- Slices come off the agarose as they are being cut in the vibratome: Ensure that a good interface is made when embedding the brains in the low melting point agarose.
