1. Preparation of Coverslips, Culture Medium, and Digestion Enzymes
- The 12-mm round #1 glass coverslips are used for the neuronal culture. The coverslips are cleaned with 10% HCL overnight followed by ultrasonic wash with distilled and deionized water for 3 times (20 min/time). The cleaned coverslips are stored in 70% ethanol for future use. Before each experiments, the coverslips are air dried and placed into the culture plate.
- To coat the dried coverslips, 100 μl of coating solution containing 100 μg/ml Poly-D-Lysine and 10 μg/ml Laminin working solution is added onto each coverslip and the culture plate is transferred into a 37 °C incubator. After 1-2 hr, the coating solution is removed and the coverslips are washed 3 times with sterile 1X PBS.
- To prepare the culture medium, the Minimum Essential Medium (MEM) is supplemented with 5% fetal bovine serum (FBS), 1X Penicillin-Streptomycin solution (500 units of penicillin and 500 μg of streptomycin), 1X GlutaMAX-I supplement, and the antimitotic reagents containing 20 μM 5-fluoro-2-deoxyuridine and 20 μM uridine (FDU/R). For the serum-free medium, the FBS is replaced with the supplement B27.
- The collagenase A solution is prepared by dissolving collagenase A powder with MEM to make a working concentration of 1 mg/ml. For the trypsin, the 1X TrypLE Express is used.
2. Dissection and Harvest of Adult Mouse DRG Neurons
- After euthanizing the 6- to 10-weeks old adult mouse, remove the dorsal skin and cut off the whole spinal column. Wash the removed spinal column with 1 X PBS 2-3 times.
- Pin the removed spinal column to the dissecting plate with ventral side up, and carefully removes muscles to expose the sensory nerves under a dissecting microscope. The nerves connected to DRGs at the lumber level are the thickest and easy to locate. Therefore, for most experiments only the lumbar DRGs are harvested.
- Cut through each vertebra along the centerline with small scissors and carefully remove the intervertebral discs. Using forceps to split each vertebra to expose the spinal cord.
- To dissect out the lumber DRGs, lift up each lumber nerve with forceps and trace toward the spinal cord to locate the associated lumbar DRGs (from L1 to L6).
- Cut off each DRG from attached peripheral nerve, dorsal and ventral roots with spring scissors, and store it in microfuge tube with MEM medium placed on ice. More DRGs at different spinal levels (e.g. thoracic DRGs) can be dissected out in a similar way when necessary.
3. Digestion and Dissociation of Adult Mouse DRG Neurons
- After collecting all the dissected DRGs, replace the MEM medium with 1 ml collagenase A solution and incubate the microfuge tube at 37 °C for 90 min.
- Replace collagenase A solution with 500 μl 1X TrypLE Express solution and incubate at 37 °C for 15-20 min.
- Remove the TrypLE Express solution and wash the DRGs with 1 ml prepared culture medium (containing 5% FBS) for 3 times.
- Add 600 μl culture medium and gently pipette up and down to triturate the tissues for 20-30 times using a 1 ml blue, graduated pipette tip.
- After trituration, allow the non-dissociated tissues to settle down to the bottom of the microfuge and transfer the cell suspension to a 10 ml sterile tube. Add another 600 μl culture medium and repeat the trituration step until most tissues are dissociated. The obtained cell suspension contains both neurons and non-neuronal cells. In most cases, cells from 6 DRGs (~5 x104) are used for one electroporation reaction.
4. Genetic Manipulation of Neurons via Electroporation
- Prepare the transfection solution by mixing the Amaxa nucleofection solution for mouse neurons with DNA plasmids (~10 μg) or siRNA oligos (~0.2 nmol) to make a final volume of 100 μl for each transfection.
- Centrifuge the dissociated cells at 680 rpm for 7 min at room temperature, and discard supernatant as much as possible. Add the prepared transfection solution and gently pipette up and down 3-4 times with 200 μl pipette tips to re-suspend the cells.
- Transfer the cell suspension mixed with the transfection solution to the electroporation cuvette and electroporate the cells using the Amaxa Nucleofector system program G-013.
- After electroporation, immediately add 500 μl of pre-warmed (37 °C) culture medium containing FBS to the cuvette and transfer all the solution (~600 μl) into the coated culture plate at desired cell densities. For experiment that requires re-suspension and re-plating, neurons are cultured directly on the plastic culture dish at high density (10000-20000 cells/well). For experiment that directly examines axon growth, neurons are plated onto coated glass coverslips at lower density (3000-5000 cells/well). Place the culture plate into the incubator (37 °C, 5% CO2).
- Four hr after plating when neurons have attached to the substrates, gently replace the culture medium (which contains the Amaxa nucleofection solution) with 500 μl fresh and pre-warmed culture medium, and return the plate to the incubator for additional culture (37 °C, 5% CO2). Both culture medium containing FBS or the serum-free medium can be used at this step.
5. Culturing Adult DRG Neurons for Axon Growth Analysis
- For neurons transfected with DNA plasmids, the expression of gene-of-interest (e.g. EGFP) can be observed as early as a few hr after electroporation. For neurons transfected with siRNAs, we usually wait 3-4 days to allow sufficient depletion of the endogenous proteins. The cultured neurons can be either directly fixed for axon growth analysis at various time points (1-4 days after electroporation) or re-suspended and re-plated to analyze axon regrowth (see below).
- For RNAi-mediated loss-of-function studies, the cultured neurons can be re-suspended and re-plated to allow axons to regrow from neurons, in which the targeted proteins are already depleted. To do so, replace the old culture medium of the high-density cultured neurons with 1 ml pre-warmed fresh medium and pipette gently up and down for 6-10 times to re-suspend the attached neurons from the culture plate. Because non-neuronal cells (e.g. Schwann cells) attach much tighter to the culture dish than neurons, the re-suspended cells are mostly neurons.
- Transfer the re-suspended neurons into a microfuge tube and gently triturate 10-15 times to re-dissociate the cell clumps into the single cell suspension.
- Re-plate the re-dissociated neurons onto newly prepared coverslips at low density (3000-5000 cells/well) and culture overnight (16-24 hr).
6. Fixation, Immuno-staining, and Fluorescence Imaging
- Aspirate the medium and add 4% paraformaldehyde (PFA) (200 μl/well) to fix the cells for 15-20 min at room temperature. The fixed cells are then washed with 1X PBS for 3 times.
- Aspirate the PBS and add the blocking solution (1% bovine serum albumin, 0.1% Triton X-100, and 2.0% normal goat serum in 1X PBS) to the fixed neurons for 60 min at room temperature.
- To label axons, the neurons are immuno-stained with anti-neurofilaments or anti-βIII tubulin antibody. To do so, place a 30μl drop of primary antibody solution (1:1200 dilution for the -βIII tubulin antibody) on a parafilm for each coverslip. Invert the coverslips and place them onto the primary antibody solution for 60 min at room temperature.
- Return coverslips to the original culture plate and wash them with 1X PBS for 3 times.
- Repeat the same procedure for the secondary antibody.
- Wash the coverslips with distilled water for 3 times, and then mount coverslips on glass slides with the mounting solution (e.g. ProLong Gold Antifade).
- The stained neurons can be imaged with any fluorescence microscopy system equipped with a digital camera. The axon lengths are measured and analyzed with imaging analysis software.
7. Representative Results
In the absence of any added extracellular growth factors, the adult DRG neurons usually start to grow axons 48 hr after first plating. The axons often show branched morphologies (Figure 1). In contrast, the re-plated neurons start to extend axons only a few hr after plating, and the axons elongate with much reduced branching (Figure 2). These results suggest that re-plated neurons share similar properties to those of conditioning lesioned neurons. By using this approach, we have recently performed loss-of-function studies to examine the role of axon regeneration associated transcription factor c-Jun in axon growth from adult DRG neurons in vitro. The results showed that electroporation of a group of 4 different siRNAs targeting different regions of c-Jun (ON-TARGETplus) markedly reduced the protein levels of c-Jun in adult DRG neurons 3 days after transfection (Figure 3)9. When the neurons were re-plated and cultured overnight, axon growth from c-Jun knockdown neurons was significantly reduced (Control: 348.37± 16.21mm; si-c-Jun: 262.32±15.69 μm, Figure 3)9. These results indicate that cultured adult DRG neurons provide a useful model system to study axon growth from adult neurons.
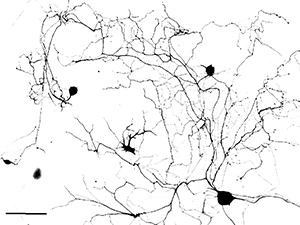
Figure 1. Adult mouse DRG neurons cultured at low density for 3 days. The neurons were stained with anti–βIII tubulin antibody. Note that most axons show branched morphologies. Scale bar: 125 μm.
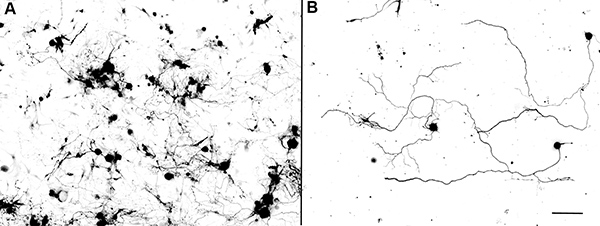
Figure 2. Re-plating adult mouse DRG neurons after 3 days in culture. (A) Adult DRG neurons cultured at high density for 3 days. (B) Re-plated adult DRG neurons were cultured at low density for overnight. Note that most axon show elongated morphologies with little axon branching. Scale bar: 250 μm in A and 125 μm in B.
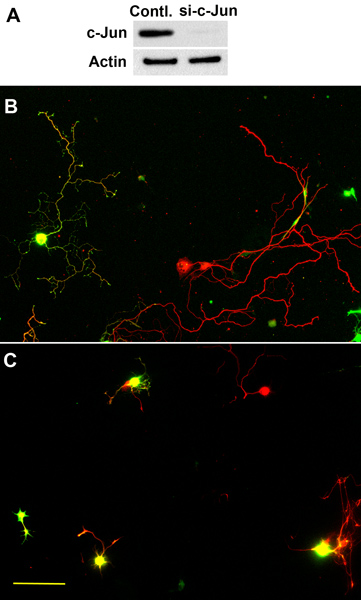
Figure 3. Role of c-Jun in axon growth from adult DRG neurons in vitro. (A) Western blot analysis of c-Jun in adult mouse DRG neurons after electroporation of c-Jun siRNAs. The result shows markedly reduced level of c-Jun. (B) Control neurons transfected with EGFP grew long axons after re-plating and overnight culture. (C) Co-transfection of c-Jun siRNAs and EGFP impaired axon growth from adult DRG neurons after re-plating and overnight culture. Red: Tuj-1 staining; Green: EGFP. Scale bar: 125 μm. These results have been published in Saijilafu et al.9.
